YAML specification
The YAML specification defines which analysis should be performed by immuneML. It is defined under three main keywords:
definitions- describing the settings ofdatasets,encodings,ml_methods,preprocessing_sequences,reports,simulationsand other components,
instructions- describing the parameters of the analysis that will be performed and which of the analysis components (defined underdefinitions) will be used for this
output- describing how to format the results of the analysis (currently, only HTML output is supported).
The purpose of this page is to list all the YAML specification options. If you are not familiar with the YAML specification and get started, see How to specify an analysis with YAML.
The overall structure of the YAML specification is the following:
definitions: # mandatory keyword
datasets: # mandatory keyword
my_dataset_1: # user-defined name of the dataset
... # see below for the specification of the dataset
encodings: # optional keyword - present if encodings are used
my_encoding_1: # user-defined name of the encoding
... # see below for the specification of different encodings
ml_methods: # optional keyword - present if ML methods are used
my_ml_method_1: # user-defined name of the ML method
ml_method_class_name: # see below for the specification of different ML methods
... # parameters of the method if any (if none are specified, default values are used)
# the parameters model_selection_cv and model_selection_n_folds can be specified for any ML method used and define if there will be
# an internal cross-validation for the given method (if used with TrainMLModel instruction, this will result in the third nested CV, but only over method parameters)
model_selection_cv: False # whether to use cross-validation and random search to estimate the optimal parameters for one split to train/test (True/False)
model_selection_n_folds: -1 # number of folds if cross-validation is used for model selection and optimal parameter estimation
preprocessing_sequences: # optional keyword - present if preprocessing sequences are used
my_preprocessing: # user-defined name of the preprocessing sequence
... # see below for the specification of different preprocessing
reports: # optional keyword - present if reports are used
my_report_1:
... # see below for the specification of different reports
instructions: # mandatory keyword - at least one instruction has to be specified
my_instruction_1: # user-defined name of the instruction
... # see below for the specification of different instructions
output: # how to present the result after running (the only valid option now)
format: HTML
A diagram of the different dataset types, preprocessing steps, encodings, ML methods and reports, and how they can be combined in different analyses is shown below. The solid lines represent components that should be used together, and the dashed lines indicate optional combinations.
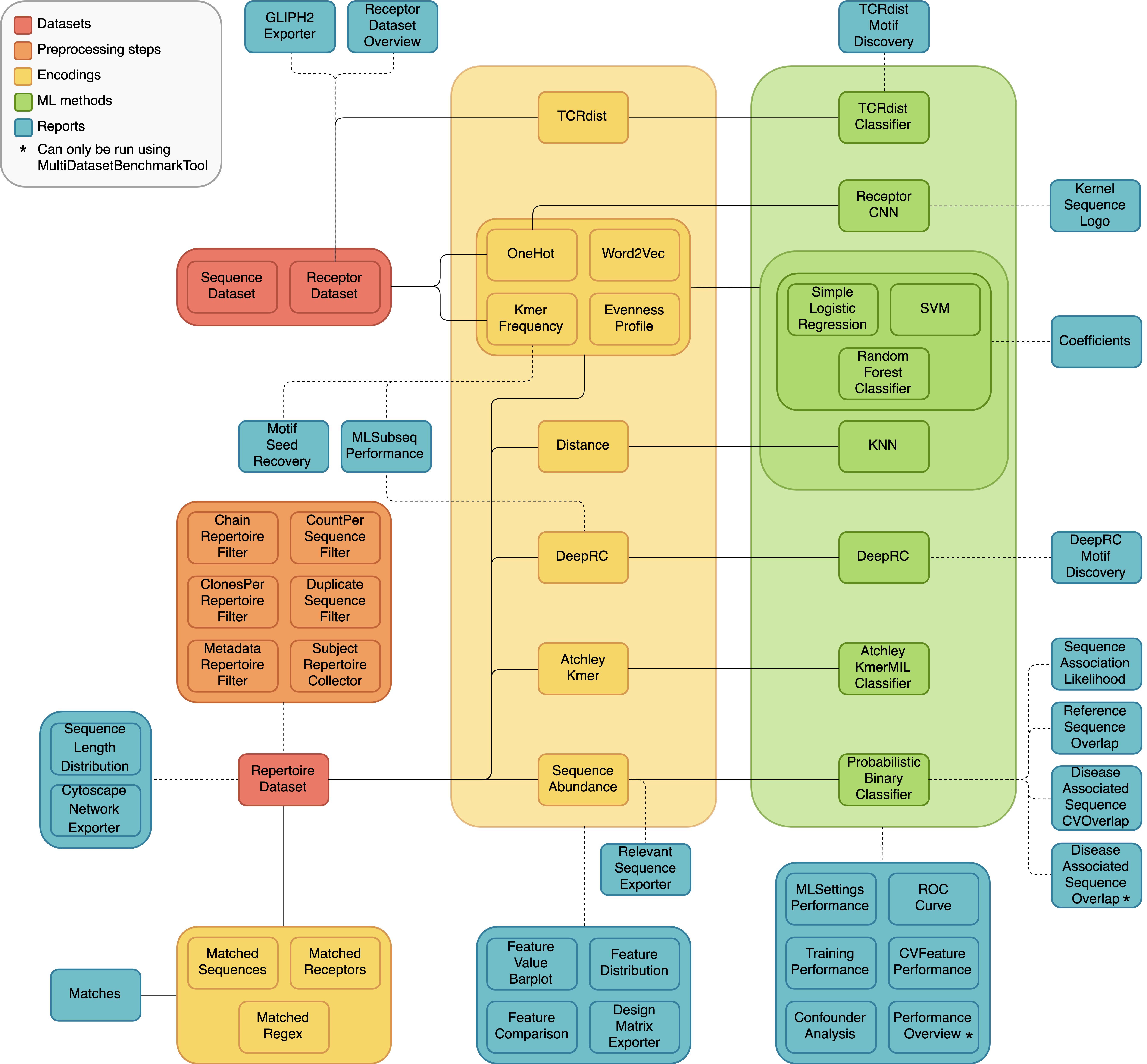
Definitions
Datasets
AIRR
Imports data in AIRR format into a Repertoire-, Sequence- or ReceptorDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets or ReceptorDatasets should be used when predicting values for unpaired (single-chain) and paired immune receptors respectively, like antigen specificity.
The AIRR .tsv format is explained here: https://docs.airr-community.org/en/stable/datarep/format.html And the AIRR rearrangement schema can be found here: https://docs.airr-community.org/en/stable/datarep/rearrangements.html
When importing a ReceptorDataset, the AIRR field cell_id is used to determine the chain pairs.
Arguments:
path (str): This is the path to a directory with AIRR files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset or ReceptorDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. Only the AIRR files included under the column ‘filename’ are imported into the RepertoireDataset. For setting SequenceDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
paired (str): Required for Sequence- or ReceptorDatasets. This parameter determines whether to import a SequenceDataset (paired = False) or a ReceptorDataset (paired = True). In a ReceptorDataset, two sequences with chain types specified by receptor_chains are paired together based on the identifier given in the AIRR column named ‘cell_id’.
receptor_chains (str): Required for ReceptorDatasets. Determines which pair of chains to import for each Receptor. Valid values are TRA_TRB, TRG_TRD, IGH_IGL, IGH_IGK. If receptor_chains is not provided, the chain pair is automatically detected (only one chain pair type allowed per repertoire).
import_productive (bool): Whether productive sequences (with value ‘T’ in column productive) should be included in the imported sequences. By default, import_productive is True.
import_with_stop_codon (bool): Whether sequences with stop codons (with value ‘T’ in column stop_codon) should be included in the imported sequences. This only applies if column stop_codon is present. By default, import_with_stop_codon is False.
import_out_of_frame (bool): Whether out of frame sequences (with value ‘F’ in column vj_in_frame) should be included in the imported sequences. This only applies if column vj_in_frame is present. By default, import_out_of_frame is False.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as AIRR uses the IMGT junction. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from AIRR column names to immuneML’s internal data representation. For AIRR, this is by default set to:
junction: sequences junction_aa: sequence_aas v_call: v_alleles j_call: j_alleles locus: chains duplicate_count: counts sequence_id: sequence_identifiersA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the AIRR file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For AIRR format, there is no default column_mapping_synonyms.
metadata_column_mapping (dict): Specifies metadata for Sequence- and ReceptorDatasets. This should specify a mapping similar to column_mapping where keys are AIRR column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for Sequence- and ReceptorDatasets. For AIRR format, there is no default metadata_column_mapping. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for AIRR this is by default “t”.
YAML specification:
my_airr_dataset: format: AIRR params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset metadata_column_mapping: # metadata column mapping AIRR: immuneML for Sequence- or ReceptorDatasetDataset airr_column_name1: metadata_label1 airr_column_name2: metadata_label2 import_productive: True # whether to include productive sequences in the dataset import_with_stop_codon: False # whether to include sequences with stop codon in the dataset import_out_of_frame: False # whether to include out of frame sequences in the dataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even if the `sequences` column is empty (provided that other fields are as specified here) import_empty_aa_sequences: False # remove all sequences with empty `sequence_aas` column # Optional fields with AIRR-specific defaults, only change when different behavior is required: separator: "\t" # column separator region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping AIRR: immuneML junction: sequences junction_aa: sequence_aas v_call: v_alleles j_call: j_alleles locus: chains duplicate_count: counts sequence_id: sequence_identifiers
Generic
Imports data from any tabular file into a Repertoire-, Sequence- or ReceptorDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets or ReceptorDatasets should be used when predicting values for unpaired (single-chain) and paired immune receptors respectively, like antigen specificity.
This importer works similarly to other importers, but has no predefined default values for which fields are imported, and can therefore be tailored to import data from various different tabular files with headers.
For ReceptorDatasets: this importer assumes the two receptor sequences appear on different lines in the file, and can be paired together by a common sequence identifier. If you instead want to import a ReceptorDataset from a tabular file that contains both receptor chains on one line, see SingleLineReceptor import
Arguments:
path (str): Required parameter. This is the path to a directory with files to import.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset or ReceptorDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. For setting Sequence- or ReceptorDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
paired (str): Required for Sequence- or ReceptorDatasets. This parameter determines whether to import a SequenceDataset (paired = False) or a ReceptorDataset (paired = True). In a ReceptorDataset, two sequences with chain types specified by receptor_chains are paired together based on a common identifier. This identifier should be mapped to the immuneML field ‘sequence_identifiers’ using the column_mapping.
receptor_chains (str): Required for ReceptorDatasets. Determines which pair of chains to import for each Receptor. Valid values are TRA_TRB, TRG_TRD, IGH_IGL, IGH_IGK.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed).
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. When IMGT_CDR3 is specified, immuneML assumes the IMGT junction (including leading C and trailing Y/F amino acids) is used in the input file, and the first and last amino acids will be removed from the sequences to retrieve the IMGT CDR3 sequence. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): Required for all datasets. A mapping where the keys are the column names in the input file, and the values correspond to the names used in immuneML’s internal data representation. Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’]. At least sequences (nucleotide) or sequence_aas (amino acids) must be specified, but all other fields are optional. A column mapping can look for example like this:
file_column_amino_acids: sequence_aas file_column_v_genes: v_genes file_column_j_genes: j_genes file_column_frequencies: countscolumn_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For Generic import, there is no default column_mapping_synonyms.
metadata_column_mapping (dict): Optional; specifies metadata for Sequence- and ReceptorDatasets. This is a column mapping that is formatted similarly to column_mapping, but here the values are the names that immuneML internally uses as metadata fields. These fields can subsequently be used as labels in instructions (for example labels that are used for prediction by ML methods). This column mapping could for example look like this:
file_column_antigen_specificity: antigen_specificityThe label antigen_specificity can now be used throughout immuneML. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
columns_to_load (list): Optional; specifies which columns to load from the input file. This may be useful if the input files contain many unused columns. If no value is specified, all columns are loaded.
separator (str): Required parameter. Column separator, for example “t” or “,”.
YAML specification:
my_generic_dataset: format: Generic params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset paired: False # whether to import SequenceDataset (False) or ReceptorDataset (True) when is_repertoire = False receptor_chains: TRA_TRB # what chain pair to import for a ReceptorDataset separator: "\t" # column separator import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping file: immuneML file_column_amino_acids: sequence_aas file_column_v_genes: v_genes file_column_j_genes: j_genes file_column_frequencies: counts metadata_column_mapping: # metadata column mapping file: immuneML file_column_antigen_specificity: antigen_specificity columns_to_load: # which subset of columns to load from the file - file_column_amino_acids - file_column_v_genes - file_column_j_genes - file_column_frequencies - file_column_antigen_specificity
IGoR
Imports data generated by IGoR simulations into a Repertoire-, or SequenceDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets should be used when predicting values for unpaired (single-chain) immune receptors, like antigen specificity.
Note that you should run IGoR with the –CDR3 option specified, this tool imports the generated CDR3 files. Sequences with missing anchors are not imported, meaning only sequences with value ‘1’ in the anchors_found column are imported. Nucleotide sequences are automatically translated to amino acid sequences.
Reference: Quentin Marcou, Thierry Mora, Aleksandra M. Walczak ‘High-throughput immune repertoire analysis with IGoR’. Nature Communications, (2018) doi.org/10.1038/s41467-018-02832-w.
Arguments:
path (str): This is the path to a directory with IGoR files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. Only the IGoR files included under the column ‘filename’ are imported into the RepertoireDataset. For setting SequenceDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
import_with_stop_codon (bool): Whether sequences with stop codons should be included in the imported sequences. By default, import_with_stop_codon is False.
import_out_of_frame (bool): Whether out of frame sequences (with value ‘0’ in column is_inframe) should be included in the imported sequences. By default, import_out_of_frame is False.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as IGoR uses the IMGT junction. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from IGoR column names to immuneML’s internal data representation. For IGoR, this is by default set to:
nt_CDR3: sequences seq_index: sequence_identifiersA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the IGoR file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For IGoR format, there is no default column_mapping_synonyms.
metadata_column_mapping (dict): Specifies metadata for SequenceDatasets. This should specify a mapping similar to column_mapping where keys are IGoR column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for SequenceDatasets. For IGoR format, there is no default metadata_column_mapping. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for IGoR this is by default “,”.
YAML specification:
my_igor_dataset: format: IGoR params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset (True) or a SequenceDataset (False) metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset metadata_column_mapping: # metadata column mapping IGoR: immuneML for SequenceDataset igor_column_name1: metadata_label1 igor_column_name2: metadata_label2 import_with_stop_codon: False # whether to include sequences with stop codon in the dataset import_out_of_frame: False # whether to include out of frame sequences in the dataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty # Optional fields with IGoR-specific defaults, only change when different behavior is required: separator: "," # column separator region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping IGoR: immuneML nt_CDR3: sequences seq_index: sequence_identifiers
IReceptor
Imports AIRR datasets retrieved through the iReceptor Gateway into a Repertoire-, Sequence- or ReceptorDataset. The differences between this importer and the AIRR importer are:
This importer takes in a list of .zip files, which must contain one or more AIRR tsv files, and for each AIRR file, a corresponding metadata json file must be present.
This importer does not require a metadata csv file for RepertoireDataset import, it is generated automatically from the metadata json files.
RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets or ReceptorDatasets should be used when predicting values for unpaired (single-chain) and paired immune receptors respectively, like antigen specificity.
AIRR rearrangement schema can be found here: https://docs.airr-community.org/en/stable/datarep/rearrangements.html
When importing a ReceptorDataset, the AIRR field cell_id is used to determine the chain pairs.
Arguments:
path (str): This is the path to a directory with .zip files retrieved from the iReceptor Gateway. These .zip files should include AIRR files (with .tsv extension) and corresponding metadata.json files with matching names (e.g., for my_dataset.tsv the corresponding metadata file is called my_dataset-metadata.json). The zip files must use the .zip extension.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset or ReceptorDataset. By default, is_repertoire is set to True.
paired (str): Required for Sequence- or ReceptorDatasets. This parameter determines whether to import a SequenceDataset (paired = False) or a ReceptorDataset (paired = True). In a ReceptorDataset, two sequences with chain types specified by receptor_chains are paired together based on the identifier given in the AIRR column named ‘cell_id’.
receptor_chains (str): Required for ReceptorDatasets. Determines which pair of chains to import for each Receptor. Valid values are TRA_TRB, TRG_TRD, IGH_IGL, IGH_IGK. If receptor_chains is not provided, the chain pair is automatically detected (only one chain pair type allowed per repertoire).
import_productive (bool): Whether productive sequences (with value ‘T’ in column productive) should be included in the imported sequences. By default, import_productive is True.
import_with_stop_codon (bool): Whether sequences with stop codons (with value ‘T’ in column stop_codon) should be included in the imported sequences. This only applies if column stop_codon is present. By default, import_with_stop_codon is False.
import_out_of_frame (bool): Whether out of frame sequences (with value ‘F’ in column vj_in_frame) should be included in the imported sequences. This only applies if column vj_in_frame is present. By default, import_out_of_frame is False.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as AIRR uses the IMGT junction. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from AIRR column names to immuneML’s internal data representation. For AIRR, this is by default set to:
junction: sequences junction_aa: sequence_aas v_call: v_alleles j_call: j_alleles locus: chains duplicate_count: counts sequence_id: sequence_identifiersA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the AIRR file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For AIRR format, there is no default column_mapping_synonyms.
metadata_column_mapping (dict): Specifies metadata for Sequence- and ReceptorDatasets. This should specify a mapping similar to column_mapping where keys are AIRR column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for Sequence- and ReceptorDatasets. For AIRR format, there is no default metadata_column_mapping. When importing a RepertoireDataset, the metadata is automatically extracted from the metadata json files.
separator (str): Column separator, for AIRR this is by default “t”.
YAML specification:
my_airr_dataset: format: IReceptor params: path: path/to/zipfiles/ is_repertoire: True # whether to import a RepertoireDataset metadata_column_mapping: # metadata column mapping AIRR: immuneML for Sequence- or ReceptorDatasetDataset airr_column_name1: metadata_label1 airr_column_name2: metadata_label2 import_productive: True # whether to include productive sequences in the dataset import_with_stop_codon: False # whether to include sequences with stop codon in the dataset import_out_of_frame: False # whether to include out of frame sequences in the dataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even if the `sequences` column is empty (provided that other fields are as specified here) import_empty_aa_sequences: False # remove all sequences with empty `sequence_aas` column # Optional fields with AIRR-specific defaults, only change when different behavior is required: separator: "\t" # column separator region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping AIRR: immuneML junction: sequences junction_aa: sequence_aas v_call: v_alleles j_call: j_alleles locus: chains duplicate_count: counts sequence_id: sequence_identifiers
ImmuneML
Imports the dataset from the files previously exported by immuneML. It closely resembles AIRR format but relies on binary representations and is optimized for faster read-in at runtime.
ImmuneMLImport can import any kind of dataset (RepertoireDataset, SequenceDataset, ReceptorDataset).
This format includes:
a dataset file in yaml format with iml_dataset extension with parameters:
name,
identifier,
metadata_file (for repertoire datasets),
metadata_fields (for repertoire datasets),
repertoire_ids (for repertoire datasets)
element_ids (for receptor and sequence datasets),
labels,
a csv metadata file (only for repertoire datasets, should be in the same folder as the iml_dataset file),
3. data files for different types of data. For repertoire datasets, data files include one binary numpy file per repertoire with sequences and associated information and one metadata yaml file per repertoire with details such as repertoire identifier, disease status, subject id and other similar available information. For sequence and receptor datasets, sequences or receptors respectively, are stored in batches in binary numpy files.
Arguments:
path (str): The path to the previously created dataset file. This file should have an ‘.iml_dataset’ extension. If the path has not been specified, immuneML attempts to load the dataset from a specified metadata file (only for RepertoireDatasets).
metadata_file (str): An optional metadata file for a RepertoireDataset. If specified, the RepertoireDataset metadata will be updated to the newly specified metadata without otherwise changing the Repertoire objects
YAML specification:
my_dataset: format: ImmuneML params: path: path/to/dataset.iml_dataset metadata_file: path/to/metadata.csv
ImmunoSEQRearrangement
Imports data from Adaptive Biotechnologies immunoSEQ Analyzer rearrangement-level .tsv files into a Repertoire-, or SequenceDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets should be used when predicting values for unpaired (single-chain) immune receptors, like antigen specificity.
The format of the files imported by this importer is described here: https://www.adaptivebiotech.com/wp-content/uploads/2019/07/MRK-00342_immunoSEQ_TechNote_DataExport_WEB_REV.pdf Alternatively, to import sample-level .tsv files, see ImmunoSEQSample import
The only difference between these two importers is which columns they load from the .tsv files.
Arguments:
path (str): This is the path to a directory with files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. Only the files included under the column ‘filename’ are imported into the RepertoireDataset. For setting SequenceDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
import_productive (bool): Whether productive sequences (with value ‘In’ in column frame_type) should be included in the imported sequences. By default, import_productive is True.
import_with_stop_codon (bool): Whether sequences with stop codons (with value ‘Stop’ in column frame_type) should be included in the imported sequences. By default, import_with_stop_codon is False.
import_out_of_frame (bool): Whether out of frame sequences (with value ‘Out’ in column frame_type) should be included in the imported sequences. By default, import_out_of_frame is False.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as immunoSEQ files use the IMGT junction. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from immunoSEQ column names to immuneML’s internal data representation. For immunoSEQ rearrangement-level files, this is by default set to:
rearrangement: sequences amino_acid: sequence_aas v_gene: v_genes j_gene: j_genes frame_type: frame_types v_family: v_subgroups j_family: j_subgroups v_allele: v_alleles j_allele: j_alleles templates: counts locus: chainsA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For immunoSEQ rearrangement-level files, this is by default set to:
v_resolved: v_alleles j_resolved: j_allelescolumns_to_load (list): Specifies which subset of columns must be loaded from the file. By default, this is: [rearrangement, v_family, v_gene, v_allele, j_family, j_gene, j_allele, amino_acid, templates, frame_type, locus]
metadata_column_mapping (dict): Specifies metadata for SequenceDatasets. This should specify a mapping similar to column_mapping where keys are immunoSEQ column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for SequenceDatasets. For immunoSEQ rearrangement .tsv files, there is no default metadata_column_mapping. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for ImmunoSEQ files this is by default “t”.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter will typically be False (import only non-empty amino acid sequences)
YAML specification:
my_immunoseq_dataset: format: ImmunoSEQRearrangement params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset (True) or a SequenceDataset (False) metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset metadata_column_mapping: # metadata column mapping ImmunoSEQ: immuneML for SequenceDataset immunoseq_column_name1: metadata_label1 immunoseq_column_name2: metadata_label2 import_productive: True # whether to include productive sequences in the dataset import_with_stop_codon: False # whether to include sequences with stop codon in the dataset import_out_of_frame: False # whether to include out of frame sequences in the dataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set # Optional fields with ImmunoSEQ rearrangement-specific defaults, only change when different behavior is required: separator: "\t" # column separator columns_to_load: # subset of columns to load - rearrangement - v_family - v_gene - v_allele - j_family - j_gene - j_allele - amino_acid - templates - frame_type - locus region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping immunoSEQ: immuneML rearrangement: sequences amino_acid: sequence_aas v_gene: v_genes j_gene: j_genes frame_type: frame_types v_family: v_subgroups j_family: j_subgroups v_allele: v_alleles j_allele: j_alleles templates: counts locus: chains
ImmunoSEQSample
Imports data from Adaptive Biotechnologies immunoSEQ Analyzer sample-level .tsv files into a Repertoire-, or SequenceDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets should be used when predicting values for unpaired (single-chain) immune receptors, like antigen specificity.
The format of the files imported by this importer is described here in section 3.4.13 https://clients.adaptivebiotech.com/assets/downloads/immunoSEQ_AnalyzerManual.pdf Alternatively, to import rearrangement-level .tsv files, see ImmunoSEQRearrangement import. The only difference between these two importers is which columns they load from the .tsv files.
Arguments:
path (str): This is the path to a directory with files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. Only the files included under the column ‘filename’ are imported into the RepertoireDataset. For setting SequenceDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
import_productive (bool): Whether productive sequences (with value ‘In’ in column frame_type) should be included in the imported sequences. By default, import_productive is True.
import_with_stop_codon (bool): Whether sequences with stop codons (with value ‘Stop’ in column frame_type) should be included in the imported sequences. By default, import_with_stop_codon is False.
import_out_of_frame (bool): Whether out of frame sequences (with value ‘Out’ in column frame_type) should be included in the imported sequences. By default, import_out_of_frame is False.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as immunoSEQ files use the IMGT junction. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from immunoSEQ column names to immuneML’s internal data representation. For immunoSEQ sample-level files, this is by default set to:
nucleotide: sequences aminoAcid: sequence_aas vGeneName: v_genes jGeneName: j_genes sequenceStatus: frame_types vFamilyName: v_subgroups jFamilyName: j_subgroups vGeneAllele: v_alleles jGeneAllele: j_alleles count (templates/reads): countsA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For immunoSEQ sample .tsv files, there is no default column_mapping_synonyms.
columns_to_load (list): Specifies which subset of columns must be loaded from the file. By default, this is: [nucleotide, aminoAcid, count (templates/reads), vFamilyName, vGeneName, vGeneAllele, jFamilyName, jGeneName, jGeneAllele, sequenceStatus]; these are the columns from the original file that will be imported
metadata_column_mapping (dict): Specifies metadata for SequenceDatasets. This should specify a mapping similar to column_mapping where keys are immunoSEQ column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for SequenceDatasets. For immunoSEQ sample .tsv files, there is no default metadata_column_mapping. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for ImmunoSEQ files this is by default “t”.
YAML specification:
my_immunoseq_dataset: format: ImmunoSEQSample params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset (True) or a SequenceDataset (False) metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset metadata_column_mapping: # metadata column mapping ImmunoSEQ: immuneML for SequenceDataset immunoseq_column_name1: metadata_label1 immunoseq_column_name2: metadata_label2 import_productive: True # whether to include productive sequences in the dataset import_with_stop_codon: False # whether to include sequences with stop codon in the dataset import_out_of_frame: False # whether to include out of frame sequences in the dataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set # Optional fields with ImmunoSEQ sample-specific defaults, only change when different behavior is required: separator: "\t" # column separator columns_to_load: # subset of columns to load - nucleotide - aminoAcid - count (templates/reads) - vFamilyName - vGeneName - vGeneAllele - jFamilyName - jGeneName - jGeneAllele - sequenceStatus region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping immunoSEQ: immuneML nucleotide: sequences aminoAcid: sequence_aas vGeneName: v_genes jGeneName: j_genes sequenceStatus: frame_types vFamilyName: v_subgroups jFamilyName: j_subgroups vGeneAllele: v_alleles jGeneAllele: j_alleles count (templates/reads): counts
MiXCR
Imports data in MiXCR format into a Repertoire-, or SequenceDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets should be used when predicting values for unpaired (single-chain) immune receptors, like antigen specificity.
Arguments:
path (str): This is the path to a directory with MiXCR files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. Only the MiXCR files included under the column ‘filename’ are imported into the RepertoireDataset. For setting SequenceDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence, such as ‘_’, are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as MiXCR uses IMGT junction as CDR3. Alternatively to importing the CDR3 sequence, other region types can be specified here as well. Valid values are IMGT_CDR3, IMGT_CDR1, IMGT_CDR2, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4.
column_mapping (dict): A mapping from MiXCR column names to immuneML’s internal data representation. For MiXCR, this is by default set to:
cloneCount: counts allVHitsWithScore: v_alleles allJHitsWithScore: j_allelesThe columns that specify the sequences to import are handled by the region_type parameter. A custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the MiXCR file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For MiXCR format, there is no default column_mapping_synonyms.
columns_to_load (list): Specifies which subset of columns must be loaded from the MiXCR file. By default, this is: [cloneCount, allVHitsWithScore, allJHitsWithScore, aaSeqCDR3, nSeqCDR3]
metadata_column_mapping (dict): Specifies metadata for SequenceDatasets. This should specify a mapping similar to column_mapping where keys are MiXCR column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for SequenceDatasets. For MiXCR format, there is no default metadata_column_mapping. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for MiXCR this is by default “t”.
YAML specification:
my_mixcr_dataset: format: MiXCR params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset (True) or a SequenceDataset (False) metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset metadata_column_mapping: # metadata column mapping MiXCR: immuneML for SequenceDataset mixcrColumnName1: metadata_label1 mixcrColumnName2: metadata_label2 region_type: IMGT_CDR3 # what part of the sequence to import import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set # Optional fields with MiXCR-specific defaults, only change when different behavior is required: separator: "\t" # column separator columns_to_load: # subset of columns to load, sequence columns are handled by region_type parameter - cloneCount - allVHitsWithScore - allJHitsWithScore - aaSeqCDR3 - nSeqCDR3 column_mapping: # column mapping MiXCR: immuneML cloneCount: counts allVHitsWithScore: v_genes allJHitsWithScore: j_genes
OLGA
Imports data generated by OLGA simulations into a Repertoire-, or SequenceDataset. Assumes that the columns in each file correspond to: nucleotide sequences, amino acid sequences, v genes, j genes
Reference: Sethna, Zachary et al. ‘High-throughput immune repertoire analysis with IGoR’. Bioinformatics, (2019) doi.org/10.1093/bioinformatics/btz035.
Arguments:
path (str): This is the path to a directory with OLGA files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. Only the OLGA files included under the column ‘filename’ are imported into the RepertoireDataset. SequenceDataset metadata is currently not supported.
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as OLGA uses the IMGT junction. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
separator (str): Column separator, for OLGA this is by default “t”.
YAML specification:
my_olga_dataset: format: OLGA params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset (True) or a SequenceDataset (False) metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set # Optional fields with OLGA-specific defaults, only change when different behavior is required: separator: "\t" # column separator region_type: IMGT_CDR3 # what part of the sequence to import
RandomReceptorDataset
Returns a ReceptorDataset consisting of randomly generated sequences, which can be used for benchmarking purposes. The sequences consist of uniformly chosen amino acids or nucleotides.
Arguments:
receptor_count (int): The number of receptors the ReceptorDataset should contain.
chain_1_length_probabilities (dict): A mapping where the keys correspond to different sequence lengths for chain 1, and the values are the probabilities for choosing each sequence length. For example, to create a random ReceptorDataset where 40% of the sequences for chain 1 would be of length 10, and 60% of the sequences would have length 12, this mapping would need to be specified:
10: 0.4 12: 0.6chain_2_length_probabilities (dict): Same as chain_1_length_probabilities, but for chain 2.
labels (dict): A mapping that specifies randomly chosen labels to be assigned to the receptors. One or multiple labels can be specified here. The keys of this mapping are the labels, and the values consist of another mapping between label classes and their probabilities. For example, to create a random ReceptorDataset with the label cmv_epitope where 70% of the receptors has class binding and the remaining 30% has class not_binding, the following mapping should be specified:
cmv_epitope: binding: 0.7 not_binding: 0.3YAML specification:
my_random_dataset: format: RandomReceptorDataset params: receptor_count: 100 # number of random receptors to generate chain_1_length_probabilities: 14: 0.8 # 80% of all generated sequences for all receptors (for chain 1) will have length 14 15: 0.2 # 20% of all generated sequences across all receptors (for chain 1) will have length 15 chain_2_length_probabilities: 14: 0.8 # 80% of all generated sequences for all receptors (for chain 2) will have length 14 15: 0.2 # 20% of all generated sequences across all receptors (for chain 2) will have length 15 labels: epitope1: # label name True: 0.5 # 50% of the receptors will have class True False: 0.5 # 50% of the receptors will have class False epitope2: # next label with classes that will be assigned to receptors independently of the previous label or other parameters 1: 0.3 # 30% of the generated receptors will have class 1 0: 0.7 # 70% of the generated receptors will have class 0
RandomRepertoireDataset
Returns a RepertoireDataset consisting of randomly generated sequences, which can be used for benchmarking purposes. The sequences consist of uniformly chosen amino acids or nucleotides.
Arguments:
repertoire_count (int): The number of repertoires the RepertoireDataset should contain.
sequence_count_probabilities (dict): A mapping where the keys are the number of sequences per repertoire, and the values are the probabilities that any of the repertoires would have that number of sequences. For example, to create a random RepertoireDataset where 40% of the repertoires would have 1000 sequences, and the other 60% would have 1100 sequences, this mapping would need to be specified:
1000: 0.4 1100: 0.6sequence_length_probabilities (dict): A mapping where the keys correspond to different sequence lengths, and the values are the probabilities for choosing each sequence length. For example, to create a random RepertoireDataset where 40% of the sequences would be of length 10, and 60% of the sequences would have length 12, this mapping would need to be specified:
10: 0.4 12: 0.6labels (dict): A mapping that specifies randomly chosen labels to be assigned to the Repertoires. One or multiple labels can be specified here. The keys of this mapping are the labels, and the values consist of another mapping between label classes and their probabilities. For example, to create a random RepertoireDataset with the label CMV where 70% of the Repertoires has class cmv_positive and the remaining 30% has class cmv_negative, the following mapping should be specified:
CMV: cmv_positive: 0.7 cmv_negative: 0.3YAML specification:
my_random_dataset: format: RandomRepertoireDataset params: repertoire_count: 100 # number of random repertoires to generate sequence_count_probabilities: 10: 0.5 # probability that any of the repertoires would have 10 receptor sequences 20: 0.5 sequence_length_probabilities: 10: 0.5 # probability that any of the receptor sequences would be 10 amino acids in length 12: 0.5 labels: # randomly assigned labels (only useful for simple benchmarking) cmv: True: 0.5 # probability of value True for label cmv to be assigned to any repertoire False: 0.5
RandomSequenceDataset
Returns a SequenceDataset consisting of randomly generated sequences, which can be used for benchmarking purposes. The sequences consist of uniformly chosen amino acids or nucleotides.
Arguments:
sequence_count (int): The number of sequences the SequenceDataset should contain.
length_probabilities (dict): A mapping where the keys correspond to different sequence lengths and the values are the probabilities for choosing each sequence length. For example, to create a random SequenceDataset where 40% of the sequences would be of length 10, and 60% of the sequences would have length 12, this mapping would need to be specified:
10: 0.4 12: 0.6labels (dict): A mapping that specifies randomly chosen labels to be assigned to the sequences. One or multiple labels can be specified here. The keys of this mapping are the labels, and the values consist of another mapping between label classes and their probabilities. For example, to create a random SequenceDataset with the label cmv_epitope where 70% of the sequences has class binding and the remaining 30% has class not_binding, the following mapping should be specified:
cmv_epitope: binding: 0.7 not_binding: 0.3YAML specification:
my_random_dataset: format: RandomSequenceDataset params: sequence_count: 100 # number of random sequences to generate length_probabilities: 14: 0.8 # 80% of all generated sequences for all sequences will have length 14 15: 0.2 # 20% of all generated sequences across all sequences will have length 15 labels: epitope1: # label name True: 0.5 # 50% of the sequences will have class True False: 0.5 # 50% of the sequences will have class False epitope2: # next label with classes that will be assigned to sequences independently of the previous label or other parameters 1: 0.3 # 30% of the generated sequences will have class 1 0: 0.7 # 70% of the generated sequences will have class 0
SingleLineReceptor
Imports data from a tabular file (where each line contains a pair of immune receptor sequences) into a ReceptorDataset. If you instead want to import a ReceptorDataset from a tabular file that contains one receptor sequence per line, see Generic import.
Arguments:
path (str): Required parameter. This is the path to a directory with files to import.
receptor_chains (str): Required parameter. Determines which pair of chains to import for each Receptor. Valid values for receptor_chains are: TRA_TRB, TRG_TRD, IGH_IGL, IGH_IGK.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. When IMGT_CDR3 is specified, immuneML assumes the IMGT junction (including leading C and trailing Y/F amino acids) is used in the input file, and the first and last amino acids will be removed from the sequences to retrieve the IMGT CDR3 sequence. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping where the keys are the column names in the input file, and the values must be mapped to the following fields: <chain>_amino_acid_sequence, <chain>_nucleotide_sequence, <chain>_v_gene, <chain>_j_gene, identifier, epitope. The possible names that can be filled in for <chain> are: ALPHA, BETA, GAMMA, DELTA, HEAVY, LIGHT, KAPPA. Any column namme other than the sequence, v/j genes and identifier will be set as metadata fields to the Receptors, and can subsequently be used as labels in immuneML instructions. For TCR alpha-beta receptor import, a column mapping could for example look like this:
cdr3_a_aa: alpha_amino_acid_sequence cdr3_b_aa: beta_amino_acid_sequence cdr3_a_nucseq: alpha_nucleotide_sequence cdr3_b_nucseq: beta_nucleotide_sequence v_a_gene: alpha_v_gene v_b_gene: beta_v_gene j_a_gene: alpha_j_gene j_b_gene: beta_j_gene clone_id: identifier epitope: epitope # metadata fieldcolumn_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded.
columns_to_load (list): Optional; specifies which columns to load from the input file. This may be useful if the input files contain many unused columns. If no value is specified, all columns are loaded.
separator (str): Required parameter. Column separator, for example “t” or “,”.
organism (str): The organism that the receptors came from. This will be set as a parameter in the ReceptorDataset object.
YAML specification:
my_receptor_dataset: format: SingleLineReceptor params: path: path/to/files/ receptor_chains: TRA_TRB # what chain pair to import separator: "\t" # column separator import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set region_type: IMGT_CDR3 # what part of the sequence to import columns_to_load: # which subset of columns to load from the file - subject - epitope - count - v_a_gene - j_a_gene - cdr3_a_aa - v_b_gene - j_b_gene - cdr3_b_aa - clone_id column_mapping: # column mapping file: immuneML cdr3_a_aa: alpha_amino_acid_sequence cdr3_b_aa: beta_amino_acid_sequence cdr3_a_nucseq: alpha_nucleotide_sequence cdr3_b_nucseq: beta_nucleotide_sequence v_a_gene: alpha_v_gene v_b_gene: beta_v_gene j_a_gene: alpha_j_gene j_b_gene: beta_j_gene clone_id: identifier epitope: epitope organism: mouse
TenxGenomics
Imports data from the 10x Genomics Cell Ranger analysis pipeline into a Repertoire-, Sequence- or ReceptorDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets or ReceptorDatasets should be used when predicting values for unpaired (single-chain) and paired immune receptors respectively, like antigen specificity.
The files that should be used as input are named ‘Clonotype consensus annotations (CSV)’, as described here: https://support.10xgenomics.com/single-cell-vdj/software/pipelines/latest/output/annotation#consensus
Note: by default the 10xGenomics field ‘umis’ is used to define the immuneML field counts. If you want to use the 10x Genomics field reads instead, this can be changed in the column_mapping (set reads: counts). Furthermore, the 10xGenomics field clonotype_id is used for the immuneML field cell_id.
Arguments:
path (str): This is the path to a directory with 10xGenomics files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset or ReceptorDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. For setting Sequence- or ReceptorDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
paired (str): Required for Sequence- or ReceptorDatasets. This parameter determines whether to import a SequenceDataset (paired = False) or a ReceptorDataset (paired = True). In a ReceptorDataset, two sequences with chain types specified by receptor_chains are paired together based on the identifier given in the 10xGenomics column named ‘clonotype_id’.
receptor_chains (str): Required for ReceptorDatasets. Determines which pair of chains to import for each Receptor. Valid values are TRA_TRB, TRG_TRD, IGH_IGL, IGH_IGK. If receptor_chains is not provided, the chain pair is automatically detected (only one chain pair type allowed per repertoire).
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as 10xGenomics uses IMGT junction as CDR3. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from 10xGenomics column names to immuneML’s internal data representation. For 10xGenomics, this is by default set to:
cdr3: sequence_aas cdr3_nt: sequences v_gene: v_genes j_gene: j_genes umis: counts chain: chains clonotype_id: cell_ids consensus_id: sequence_identifiersA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the 10xGenomics file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For 10xGenomics format, there is no default column_mapping_synonyms.
metadata_column_mapping (dict): Specifies metadata for Sequence- and ReceptorDatasets. This should specify a mapping similar to column_mapping where keys are 10xGenomics column names and values are the names that are internally used in immuneML as metadata fields. These metadata fields can be used as prediction labels for Sequence- and ReceptorDatasets. For 10xGenomics format, there is no default metadata_column_mapping. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for 10xGenomics this is by default “,”.
YAML specification:
my_10x_dataset: format: 10xGenomics params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset paired: False # whether to import SequenceDataset (False) or ReceptorDataset (True) when is_repertoire = False receptor_chains: TRA_TRB # what chain pair to import for a ReceptorDataset metadata_column_mapping: # metadata column mapping 10xGenomics: immuneML for SequenceDataset tenx_column_name1: metadata_label1 tenx_column_name2: metadata_label2 import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set # Optional fields with 10xGenomics-specific defaults, only change when different behavior is required: separator: "," # column separator region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping 10xGenomics: immuneML cdr3: sequence_aas cdr3_nt: sequences v_gene: v_genes j_gene: j_genes umis: counts chain: chains clonotype_id: cell_ids consensus_id: sequence_identifiers
VDJdb
Imports data in VDJdb format into a Repertoire-, Sequence- or ReceptorDataset. RepertoireDatasets should be used when making predictions per repertoire, such as predicting a disease state. SequenceDatasets or ReceptorDatasets should be used when predicting values for unpaired (single-chain) and paired immune receptors respectively, like antigen specificity.
Arguments:
path (str): This is the path to a directory with VDJdb files to import. By default path is set to the current working directory.
is_repertoire (bool): If True, this imports a RepertoireDataset. If False, it imports a SequenceDataset or ReceptorDataset. By default, is_repertoire is set to True.
metadata_file (str): Required for RepertoireDatasets. This parameter specifies the path to the metadata file. This is a csv file with columns filename, subject_id and arbitrary other columns which can be used as labels in instructions. For setting Sequence- or ReceptorDataset metadata, metadata_file is ignored, see metadata_column_mapping instead.
paired (str): Required for Sequence- or ReceptorDatasets. This parameter determines whether to import a SequenceDataset (paired = False) or a ReceptorDataset (paired = True). In a ReceptorDataset, two sequences with chain types specified by receptor_chains are paired together based on the identifier given in the VDJdb column named ‘complex.id’.
receptor_chains (str): Required for ReceptorDatasets. Determines which pair of chains to import for each Receptor. Valid values are TRA_TRB, TRG_TRD, IGH_IGL, IGH_IGK. If receptor_chains is not provided, the chain pair is automatically detected (only one chain pair type allowed per repertoire).
import_illegal_characters (bool): Whether to import sequences that contain illegal characters, i.e., characters that do not appear in the sequence alphabet (amino acids including stop codon ‘*’, or nucleotides). When set to false, filtering is only applied to the sequence type of interest (when running immuneML in amino acid mode, only entries with illegal characters in the amino acid sequence are removed). By default import_illegal_characters is False.
import_empty_nt_sequences (bool): imports sequences which have an empty nucleotide sequence field; can be True or False. By default, import_empty_nt_sequences is set to True.
import_empty_aa_sequences (bool): imports sequences which have an empty amino acid sequence field; can be True or False; for analysis on amino acid sequences, this parameter should be False (import only non-empty amino acid sequences). By default, import_empty_aa_sequences is set to False.
region_type (str): Which part of the sequence to import. By default, this value is set to IMGT_CDR3. This means the first and last amino acids are removed from the CDR3 sequence, as VDJdb uses IMGT junction as CDR3. Specifying any other value will result in importing the sequences as they are. Valid values are IMGT_CDR1, IMGT_CDR2, IMGT_CDR3, IMGT_FR1, IMGT_FR2, IMGT_FR3, IMGT_FR4, IMGT_JUNCTION, FULL_SEQUENCE.
column_mapping (dict): A mapping from VDJdb column names to immuneML’s internal data representation. For VDJdb, this is by default set to:
V: v_alleles J: j_alleles CDR3: sequence_aas complex.id: sequence_identifiers Gene: chainsA custom column mapping can be specified here if necessary (for example; adding additional data fields if they are present in the VDJdb file, or using alternative column names). Valid immuneML fields that can be specified here are [‘sequence_aas’, ‘sequences’, ‘v_genes’, ‘j_genes’, ‘v_subgroups’, ‘j_subgroups’, ‘v_alleles’, ‘j_alleles’, ‘chains’, ‘counts’, ‘frame_types’, ‘sequence_identifiers’, ‘cell_ids’].
column_mapping_synonyms (dict): This is a column mapping that can be used if a column could have alternative names. The formatting is the same as column_mapping. If some columns specified in column_mapping are not found in the file, the columns specified in column_mapping_synonyms are instead attempted to be loaded. For VDJdb format, there is no default column_mapping_synonyms.
metadata_column_mapping (dict): Specifies metadata for Sequence- and ReceptorDatasets. This should specify a mapping where keys are VDJdb column names and values are the names that are internally used in immuneML as metadata fields. For VDJdb format, this parameter is by default set to:
Epitope: epitope Epitope gene: epitope_gene Epitope species: epitope_speciesThis means that epitope, epitope_gene and epitope_species can be specified as prediction labels for Sequence- and ReceptorDatasets. Custom metadata labels can be defined here as well. For setting RepertoireDataset metadata, metadata_column_mapping is ignored, see metadata_file instead.
separator (str): Column separator, for VDJdb this is by default “t”.
YAML specification:
my_vdjdb_dataset: format: VDJdb params: path: path/to/files/ is_repertoire: True # whether to import a RepertoireDataset metadata_file: path/to/metadata.csv # metadata file for RepertoireDataset paired: False # whether to import SequenceDataset (False) or ReceptorDataset (True) when is_repertoire = False receptor_chains: TRA_TRB # what chain pair to import for a ReceptorDataset import_illegal_characters: False # remove sequences with illegal characters for the sequence_type being used import_empty_nt_sequences: True # keep sequences even though the nucleotide sequence might be empty import_empty_aa_sequences: False # filter out sequences if they don't have sequence_aa set # Optional fields with VDJdb-specific defaults, only change when different behavior is required: separator: "\t" # column separator region_type: IMGT_CDR3 # what part of the sequence to import column_mapping: # column mapping VDJdb: immuneML V: v_genes J: j_genes CDR3: sequence_aas complex.id: sequence_identifiers Gene: chains metadata_column_mapping: # metadata column mapping VDJdb: immuneML Epitope: epitope Epitope gene: epitope_gene Epitope species: epitope_species
Simulation
Motif
Class describing motifs where each motif is defined by a seed and a way of creating specific instances of the motif (instantiation_strategy);
When instantiation_strategy is set, specific motif instances will be produced by calling instantiate_motif(seed) method of instantiation_strategy
Arguments:
seed (str): An amino acid sequence that represents the basic motif seed. All implanted motifs correspond to the seed, or a modified version thereof, as specified in it’s instantiation strategy. If this argument is set, seed_chain1 and seed_chain2 arguments are not used.
instantiation (
MotifInstantiationStrategy): Which strategy to use for implanting the seed. Valid values are: GappedKmer. In the YAML specification this can either be one of these values as a string in which case the default parameters will be used. Alternatively, instantiation can be specified with parameters as in the example YAML specification below. For the detailed list of parameters, see the specific instantiation strategies below.seed_chain1 (str): in case when representing motifs for paired chain data, it is possible to define a motif seed per chain; if this parameter is set, the generated motif instances will include a motif instance for both chains; for more details on how it works see seed argument above. Used only if the seed argument is not set.
seed_chain2 (str): used for paired chain data, for the other receptor chain; for more details on how it works see seed argument. This argument is used only if the seed argument is not set.
name_chain1: name of the first chain if paired receptor data are simulated. The value should be an instance of
Chain. This argument is used only if the seed argument is not set.name_chain2: name of the second chain 2 if paired receptor data are simulated. The value should be an instance of
Chain. This argument is used only if the seed argument is not set.YAML specification:
motifs: # examples for single chain receptor data my_simple_motif: # this will be the identifier of the motif seed: AAA instantiation: GappedKmer my_gapped_motif: seed: AA/A instantiation: GappedKmer: min_gap: 1 max_gap: 2 # examples for paired chain receptor data my_paired_motif: seed_chain1: AAA # seed for chain1 or chain2 can optionally include gap, same as for single chain receptor data name_chain1: ALPHA # alpha chain of TCR seed_chain2: CCC name_chain2: BETA # beta chain of TCR instantiation: GappedKmer # same as for single chain receptor data
GappedKmer
Creates a motif instance from a given seed and additional optional parameters. Currently, at most a single gap can be specified in the sequence.
Arguments:
min_gap (int): The minimum gap length, in case the original seed contains a gap.
max_gap (int): The maximum gap length, in case the original seed contains a gap.
hamming_distance_probabilities (dict): The probability of modifying the given seed with each number of modifications. The keys represent the number of modifications (hamming distance) between the original seed and the implanted motif, and the values represent the probabilities for the respective number of modifications. For example {0: 0.7, 1: 0.3} means that 30% of the time one position will be modified, and the remaining 70% of the time the motif will remain unmodified with respect to the seed. The values of hamming_distance_probabilities must sum to 1.
position_weights (dict): A dictionary containing the relative probabilities of choosing each position for hamming distance modification. The keys represent the position in the seed, where counting starts at 0. If the index of a gap is specified in position_weights, it will be removed. The values represent the relative probabilities for modifying each position when it gets selected for modification. For example {0: 0.6, 1: 0, 2: 0.4} means that when a sequence is selected for a modification (as specified in hamming_distance_probabilities), then 60% of the time the amino acid at index 0 is modified, and the remaining 40% of the time the amino acid at index 2. If the values of position_weights do not sum to 1, the remainder will be redistributed over all positions, including those not specified.
alphabet_weights (dict): A dictionary describing the relative probabilities of choosing each amino acid for hamming distance modification. The keys represent the amino acids and the values the relative probabilities for choosing this amino acid. If the values of alphabet_weights do not sum to 1, the remainder will be redistributed over all possible amino acids, including those not specified.
YAML specification:
GappedKmer: min_gap: 1 max_gap: 2 hamming_distance_probabilities: 0: 0.7 1: 0.3 position_weights: # note that index 2, the position of the gap, is excluded from position_weights 0: 1 1: 0 3: 0 alphabet_weights: A: 0.2 C: 0.2 D: 0.4 E: 0.2
Signal
This class represents the signal that will be implanted during a Simulation. A signal is represented by a list of motifs, and an implanting strategy.
A signal is associated with a metadata label, which is assigned to a receptor or repertoire. For example antigen-specific/disease-associated (receptor) or diseased (repertoire).
Arguments:
motifs (list): A list of the motifs associated with this signal.
implanting (
SignalImplantingStrategy): The strategy that is used to decide in which sequences the motifs should be implanted, and how.Valid values are: FullSequence, Receptor, HealthySequence
YAML specification:
signals: my_signal: motifs: - my_simple_motif - my_gapped_motif implanting: HealthySequence sequence_position_weights: 109: 0.5 110: 0.5
FullSequence
This class represents a
SignalImplantingStrategywhere signals will be implanted in the repertoire by replacing repertoire_implanting_rate percent of the sequences with sequences generated from the motifs of the signal. Motifs here cannot include gaps and the motif instances are the full sequences and will be a part of the repertoire.Note: when providing the sequence to be implanted, check if the import setting regarding the sequence type (CDR3 vs IMGT junction) matches the sequence to be implanted. For example, if importing would convert junction sequences to CDR3, but the sequence specified here for implanting would be the junction, the results of the simulation could be inconsistent.
Arguments: this signal implanting strategy has no arguments.
YAML specification:
motifs: my_motif: # cannot include gaps ... signals: my_signal: motifs: - my_motif implanting: FullSequence
Receptor
This class represents a
SignalImplantingStrategywhere signals will be implanted in both chains of immune receptors. This class should be used only when simulating paired chain data.Arguments:
implanting: name of the implanting strategy, here Receptor
sequence_position_weights (dict): A dictionary describing the relative weights for implanting a signal at each given IMGT position in the receptor sequence. If sequence_position_weights are not set, then SequenceImplantingStrategy will make all of the positions equally likely for each receptor sequence.
YAML specification:
motifs: my_motif: ... signals: my_signal: motifs: - my_motif - ... implanting: Receptor sequence_position_weights: 109: 1 110: 2 111: 5 112: 1
HealthySequence
This class represents a
SignalImplantingStrategywhere signals will be implanted in ‘healthy sequences’, meaning sequences in which no signal has been implanted previously. This ensures that there is only one signal per receptor sequence.If for the given number of sequences in the repertoire and repertoire implanting rate, the total number of sequences for implanting turns out to be less than 1 (e.g. for 12000 sequences and repertoire implanting rate 0.00005, it should implant the signal in 0.6 sequences), the signal will not be implanted in that repertoire and a warning with repertoire identifier along with the repertoire implanting rate and number of sequences in the repertoire will be raised.
Arguments:
implanting: name of the implanting strategy, here HealthySequence
sequence_position_weights (dict): A dictionary describing the relative weights for implanting a signal at each given IMGT position in the receptor sequence. If sequence_position_weights are not set, then SequenceImplantingStrategy will make all of the positions equally likely for each receptor sequence.
implanting_computation (str): defines how to determine the number of sequences to implant the signal in a repertoire; it relies on repertoire_implanting_rate, but in case where the number of sequences for implanting is not an integer, this option can be useful. If implanting rate is set to ‘round’, then the number of sequences for implanting in a repertoire will be rounded to the nearest integer value of the product of repertoire implanting rate and the number of sequences in a repertoire (e.g., if the product value is 1.2, the signal will be implanted in one sequence only). If implanting rate is set to ‘Poisson’, the number of sequences for implanting will be sampled from the Poisson distribution with the value of the lambda parameter being repertoire implanting rate multiplied by the number of sequences in the repertoire.
YAML specification:
motifs: my_motif: ... signals: my_signal: motifs: - my_motif - ... implanting: HealthySequence implanting_computation: Poisson sequence_position_weights: 109: 1 110: 2 111: 5 112: 1
Implanting
When performing a Simulation, one or more implantings can be specified. An implanting represents a set of signals which are implanted in a RepertoireDataset with given rates.
Multiple implantings may be specified in one simulation. In this case, each implanting will only affect its own partition of the dataset, so each repertoire can only receive implanted signals from one implanting. This way, implantings can be used to ensure signals do not overlap (one implanting per signal), or to ensure signals always occur together (multiple signals per implanting).
Arguments:
signals (list): The list of Signal objects to be implanted in a subset of the repertoires in a RepertoireDataset. When multiple signals are specified, this means that all of these signals are implanted in the same repertoires in a RepertoireDataset, although they may not be implanted in the same sequences within those repertoires (this depends on the
SignalImplantingStrategy).dataset_implanting_rate (float): The proportion of repertoires in the RepertoireDataset in which the signals should be implanted. When specifying multiple implantings, the sum of all dataset_implanting_rates should not exceed 1.
repertoire_implanting_rate (float): The proportion of sequences in a Repertoire where a motif associated with one of the signals should be implanted.
is_noise (bool): indicates whether the implanting should be regarded as noise; if it is True, the signals will be implanted as specified, but the repertoire/receptor in question will have negative class.
YAML specification:
simulations: # definitions of simulations should be under key simulations in the definitions part of the specification # one simulation with multiple implanting objects, a part of definition section my_simulation: my_implanting_1: signals: - my_signal dataset_implanting_rate: 0.5 repertoire_implanting_rate: 0.25 my_implanting_2: signals: - my_signal dataset_implanting_rate: 0.2 repertoire_implanting_rate: 0.75 # a simulation where the signals is present in the negative class as well (e.g. wrong labels, or by chance) noisy_simulation: positive_class_implanting: signals: - my_signal dataset_implanting_rate: 0.5 repertoire_implanting_rate: 0.1 # 10% of the repertoire includes the signal in the positive class negative_class_implanting: signals: - my_signal is_noise: True # means that signal will be implanted, but the label will have negative class dataset_implanting_rate: 0.5 repertoire_implanting_rate: 0.01 # 1% of negative class repertoires has the signal # in case of defining implanting for paired chain immune receptor data the simulation with implanting objects would be: my_receptor_simulation: my_receptor_implanting_1: # repertoire_implanting_rate is omitted in this case, as it is not applicable signals: - my_receptor_signal dataset_implanting_rate: 0.4 # 40% of the receptors will have signal my_receptor_signal implanted and 60% will not
Encodings
AtchleyKmer
Represents a repertoire through Atchley factors and relative abundance of k-mers. Should be used in combination with the AtchleyKmerMILClassifier.
For more details, see the original publication: Ostmeyer J, Christley S, Toby IT, Cowell LG. Biophysicochemical motifs in T cell receptor sequences distinguish repertoires from tumor-infiltrating lymphocytes and adjacent healthy tissue. Cancer Res. Published online January 1, 2019:canres.2292.2018. doi:10.1158/0008-5472.CAN-18-2292 .
Note that sequences in the repertoire with length shorter than skip_first_n_aa + skip_last_n_aa + k will not be encoded.
Arguments:
k (int): k-mer length
skip_first_n_aa (int): number of amino acids to remove from the beginning of the receptor sequence
skip_last_n_aa (int): number of amino acids to remove from the end of the receptor sequence
abundance: how to compute abundance term for k-mers; valid values are RELATIVE_ABUNDANCE, TCRB_RELATIVE_ABUNDANCE.
normalize_all_features (bool): when normalizing features to have 0 mean and unit variance, this parameter indicates if the abundance feature should be included in the normalization
YAML specification:
my_encoder: AtchleyKmer: k: 4 skip_first_n_aa: 3 skip_last_n_aa: 3 abundance: RELATIVE_ABUNDANCE normalize_all_features: False
CompAIRRDistance
Encodes a given RepertoireDataset as a distance matrix, using the Morisita-Horn distance metric. Internally, CompAIRR is used for fast calculation of overlap between repertoires. This creates a pairwise distance matrix between each of the repertoires. The distance is calculated based on the number of matching receptor chain sequences between the repertoires. This matching may be defined to permit 1 or 2 mismatching amino acid/nucleotide positions and 1 indel in the sequence. Furthermore, matching may or may not include V and J gene information, and sequence frequencies may be included or ignored.
When mismatches (differences and indels) are allowed, the Morisita-Horn similarity may exceed 1. In this case, the Morisita-Horn distance (= similarity - 1) is set to 0 to avoid negative distance scores.
Arguments:
compairr_path (Path): optional path to the CompAIRR executable. If not given, it is assumed that CompAIRR has been installed such that it can be called directly on the command line with the command ‘compairr’, or that it is located at /usr/local/bin/compairr.
keep_compairr_input (bool): whether to keep the input file that was passed to CompAIRR. This may take a lot of storage space if the input dataset is large. By default the input file is not kept.
differences (int): Number of differences allowed between the sequences of two immune receptor chains, this may be between 0 and 2. By default, differences is 0.
indels (bool): Whether to allow an indel. This is only possible if differences is 1. By default, indels is False.
ignore_counts (bool): Whether to ignore the frequencies of the immune receptor chains. If False, frequencies will be included, meaning the ‘counts’ values for the receptors available in two repertoires are multiplied. If False, only the number of unique overlapping immune receptors (‘clones’) are considered. By default, ignore_counts is False.
ignore_genes (bool): Whether to ignore V and J gene information. If False, the V and J genes between two receptor chains have to match. If True, gene information is ignored. By default, ignore_genes is False.
threads (int): The number of threads to use for parallelization. Default is 8.
YAML specification:
my_distance_encoder: CompAIRRDistance: compairr_path: optional/path/to/compairr differences: 0 indels: False ignore_counts: False ignore_genes: False
CompAIRRSequenceAbundance
This encoder works similarly to the
SequenceAbundanceEncoder, but internally uses CompAIRR to accelerate core computations.This encoder represents the repertoires as vectors where:
the first element corresponds to the number of label-associated clonotypes
the second element is the total number of unique clonotypes
To determine what clonotypes (with or without matching V/J genes) are label-associated based on a statistical test. The statistical test used is Fisher’s exact test (one-sided).
Reference: Emerson, Ryan O. et al. ‘Immunosequencing Identifies Signatures of Cytomegalovirus Exposure History and HLA-Mediated Effects on the T Cell Repertoire’. Nature Genetics 49, no. 5 (May 2017): 659–65. doi.org/10.1038/ng.3822.
Note: to use this encoder, it is necessary to explicitly define the positive class for the label when defining the label in the instruction. With positive class defined, it can then be determined which sequences are indicative of the positive class. See Reproduction of the CMV status predictions study for an example using
SequenceAbundanceEncoder.Arguments:
p_value_threshold (float): The p value threshold to be used by the statistical test.
compairr_path (Path): optional path to the CompAIRR executable. If not given, it is assumed that CompAIRR has been installed such that it can be called directly on the command line with the command ‘compairr’, or that it is located at /usr/local/bin/compairr.
ignore_genes (bool): Whether to ignore V and J gene information. If False, the V and J genes between two receptor chains have to match. If True, gene information is ignored. By default, ignore_genes is False.
sequence_batch_size (int): The number of sequences in a batch when comparing sequences across repertoires, typically 100s of thousands. This does not affect the results of the encoding, only the speed and memory usage.
threads (int): The number of threads to use for parallelization. This does not affect the results of the encoding, only the speed. The default number of threads is 8.
YAML specification:
my_sa_encoding: CompAIRRSequenceAbundance: compairr_path: optional/path/to/compairr p_value_threshold: 0.05 ignore_genes: False threads: 8
DeepRC
DeepRCEncoder should be used in combination with the DeepRC ML method (DeepRC). This encoder writes the data in a RepertoireDataset to .tsv files. For each repertoire, one .tsv file is created containing the amino acid sequences and the counts. Additionally, one metadata .tsv file is created, which describes the subset of repertoires that is encoded by a given instance of the DeepRCEncoder.
Note: sequences where count is None, the count value will be set to 1
YAML specification:
my_deeprc_encoder: DeepRC
Distance
Encodes a given RepertoireDataset as distance matrix, where the pairwise distance between each of the repertoires is calculated. The distance is calculated based on the presence/absence of elements defined under attributes_to_match. Thus, if attributes_to_match contains only ‘sequence_aas’, this means the distance between two repertoires is maximal if they contain the same set of sequence_aas, and the distance is minimal if none of the sequence_aas are shared between two repertoires.
Arguments:
distance_metric (
DistanceMetricType): The metric used to calculate the distance between two repertoires. Valid values are: JACCARD, MORISITA_HORN. The default distance metric is JACCARD (inverse Jaccard).sequence_batch_size (int): The number of sequences to be processed at once. Increasing this number increases the memory use. The default value is 1000.
attributes_to_match (list): The attributes to consider when determining whether a sequence is present in both repertoires. Only the fields defined under attributes_to_match will be considered, all other fields are ignored. Valid values are sequence_aas, sequences, v_genes, j_genes, v_subgroups, j_subgroups, v_alleles, j_alleles, chains, counts, region_types, frame_types, sequence_identifiers, cell_ids. The default value is [‘sequence_aas’]
YAML specification:
my_distance_encoder: Distance: distance_metric: JACCARD sequence_batch_size: 1000 attributes_to_match: - sequence_aas - v_genes - j_genes - chains - region_types
EvennessProfile
The EvennessProfileEncoder class encodes a repertoire based on the clonal frequency distribution. The evenness for a given repertoire is defined as follows:
\[^{\alpha} \mathrm{E}(\mathrm{f})=\frac{\left(\sum_{\mathrm{i}=1}^{\mathrm{n}} \mathrm{f}_{\mathrm{i}}^{\alpha}\right)^{\frac{1}{1-\alpha}}}{\mathrm{n}}\]That is, it is the exponential of Renyi entropy at a given alpha divided by the species richness, or number of unique sequences.
Reference: Greiff et al. (2015). A bioinformatic framework for immune repertoire diversity profiling enables detection of immunological status. Genome Medicine, 7(1), 49. doi.org/10.1186/s13073-015-0169-8
Arguments:
min_alpha (float): minimum alpha value to use
max_alpha (float): maximum alpha value to use
dimension (int): dimension of output evenness profile vector, or the number of alpha values to linearly space between min_alpha and max_alpha
YAML specification:
my_evenness_profile: EvennessProfile: min_alpha: 0 max_alpha: 10 dimension: 51
KmerFrequency
The KmerFrequencyEncoder class encodes a repertoire, sequence or receptor by frequencies of k-mers it contains. A k-mer is a sequence of letters of length k into which an immune receptor sequence can be decomposed. K-mers can be defined in different ways, as determined by the sequence_encoding.
Arguments:
sequence_encoding (
SequenceEncodingType): The type of k-mers that are used. The simplest sequence_encoding isCONTINUOUS_KMER, which uses contiguous subsequences of length k to represent the k-mers. When gapped k-mers are used (GAPPED_KMER,GAPPED_KMER), the k-mers may contain gaps with a size between min_gap and max_gap, and the k-mer length is defined as a combination of k_left and k_right. When IMGT k-mers are used (IMGT_CONTINUOUS_KMER,IMGT_GAPPED_KMER), IMGT positional information is taken into account (i.e. the same sequence in a different position is considered to be a different k-mer). When the identity representation is used (IDENTITY), the k-mers just correspond to the original sequences.normalization_type (
NormalizationType): The way in which the k-mer frequencies should be normalized. The default value for normalization_type is l2.reads (
ReadsType): Reads type signify whether the counts of the sequences in the repertoire will be taken into account. IfUNIQUE, only unique sequences (clonotypes) are encoded, and ifALL, the sequence ‘count’ value is taken into account when determining the k-mer frequency. The default value for reads is unique.k (int): Length of the k-mer (number of amino acids) when ungapped k-mers are used. The default value for k is 3.
k_left (int): When gapped k-mers are used, k_left indicates the length of the k-mer left of the gap. The default value for k_left is 1.
k_right (int): Same as k_left, but k_right determines the length of the k-mer right of the gap. The default value for k_right is 1.
min_gap (int): Minimum gap size when gapped k-mers are used. The default value for min_gap is 0.
max_gap: (int): Maximum gap size when gapped k-mers are used. The default value for max_gap is 0.
sequence_type (
SequenceType): Whether to work with nucleotide or amino acid sequences. Amino acid sequences are the default. To work with either sequence type, the sequences of the desired type should be included in the datasets, e.g., listed under ‘columns_to_load’ parameter. By default, both types will be included if available. Valid values are: AMINO_ACID and NUCLEOTIDE.scale_to_unit_variance (bool): whether to scale the design matrix after normalization to have unit variance per feature. Setting this argument to True might improve the subsequent classifier’s performance depending on the type of the classifier. The default value for scale_to_unit_variance is true.
scale_to_zero_mean (bool): whether to scale the design matrix after normalization to have zero mean per feature. Setting this argument to True might improve the subsequent classifier’s performance depending on the type of the classifier. However, if the original design matrix was sparse, setting this argument to True will destroy the sparsity and will increase the memory consumption. The default value for scale_to_zero_mean is false.
YAML specification:
my_continuous_kmer: KmerFrequency: normalization_type: RELATIVE_FREQUENCY reads: UNIQUE sequence_encoding: CONTINUOUS_KMER sequence_type: NUCLEOTIDE k: 3 scale_to_unit_variance: True scale_to_zero_mean: True my_gapped_kmer: KmerFrequency: normalization_type: RELATIVE_FREQUENCY reads: UNIQUE sequence_encoding: GAPPED_KMER sequence_type: AMINO_ACID k_left: 2 k_right: 2 min_gap: 1 max_gap: 3 scale_to_unit_variance: True scale_to_zero_mean: False
MatchedReceptors
Encodes the dataset based on the matches between a dataset containing unpaired (single chain) data, and a paired reference receptor dataset. For each paired reference receptor, the frequency of either chain in the dataset is counted.
This encoding can be used in combination with the Matches report.
When sum_matches and normalize are set to True, this encoder behaves similarly as described in: Yao, Y. et al. ‘T cell receptor repertoire as a potential diagnostic marker for celiac disease’. Clinical Immunology Volume 222 (January 2021): 108621. doi.org/10.1016/j.clim.2020.108621 with the only exception being that this encoder uses paired receptors, while the original publication used single sequences (see also: MatchedSequences encoder).
Arguments:
reference (dict): A dictionary describing the reference dataset file. Import should be specified the same way as regular dataset import. It is only allowed to import a receptor dataset here (i.e., is_repertoire is False and paired is True by default, and these are not allowed to be changed).
max_edit_distances (dict): A dictionary specifying the maximum edit distance between a target sequence (from the repertoire) and the reference sequence. A maximum distance can be specified per chain, for example to allow for less strict matching of TCR alpha and BCR light chains. When only an integer is specified, this distance is applied to all possible chains.
reads (
ReadsType): Reads type signify whether the counts of the sequences in the repertoire will be taken into account. IfUNIQUE, only unique sequences (clonotypes) are counted, and ifALL, the sequence ‘count’ value is summed when determining the number of matches. The default value for reads is all.sum_matches (bool): When sum_matches is False, the resulting encoded data matrix contains multiple columns with the number of matches per reference receptor chain. When sum_matches is true, the columns representing each of the two chains are summed together, meaning that there are only two aggregated sums of matches (one per chain) per repertoire in the encoded data. To use this encoder in combination with the Matches report, sum_matches must be set to False. When sum_matches is set to True, this encoder behaves similarly to the encoder described by Yao, Y. et al. By default, sum_matches is False.
normalize (bool): If True, the chain matches are divided by the total number of unique receptors in the repertoire (when reads = unique) or the total number of reads in the repertoire (when reads = all).
YAML Specification:
my_mr_encoding: MatchedReceptors: reference: path: path/to/file.txt format: VDJDB max_edit_distances: alpha: 1 beta: 0
MatchedRegex
Encodes the dataset based on the matches between a RepertoireDataset and a collection of regular expressions. For each regular expression, the number of sequences in the RepertoireDataset containing the expression is counted. This can also be used to count how often a subsequence occurs in a RepertoireDataset.
The regular expressions are defined per chain, and it is possible to require a V gene match in addition to the CDR3 sequence containing the regular expression.
This encoding can be used in combination with the Matches report.
Arguments:
match_v_genes (bool): Whether V gene matches are required. If this is True, a match is only counted if the V gene matches the gene specified in the motif input file. By default match_v_genes is False.
reads (
ReadsType): Reads type signify whether the counts of the sequences in the repertoire will be taken into account. IfUNIQUE, only unique sequences (clonotypes) are counted, and ifALL, the sequence ‘count’ value is summed when determining the number of matches. The default value for reads is all.motif_filepath (str): The path to the motif input file. This should be a tab separated file containing a column named ‘id’ and for every chain that should be matched a column containing the regex (<chain>_regex) and a column containing the V gene (<chain>V) if match_v_genes is True. The chains are specified by their three letter code, valid values are: TRA, TRB, TRG, TRD, IGH, IGL, IGK.
In the simplest case, when counting the number of occurrences of a given list of k-mers in TRB sequences, the contents of the motif file could look like this:
id
TRB_regex
1
ACG
2
EDNA
3
DFWG
It is also possible to test whether paired regular expressions occur in the dataset (for example: regular expressions matching both a TRA chain and a TRB chain) by specifying them on the same line. In a more complex case where both paired and unpaired regular expressions are specified, in addition to matching the V genes, the contents of the motif file could look like this:
id
TRA_regex
TRAV
TRB_regex
TRBV
1
AGQ.GSS
TRAV35
S[APL]GQY
TRBV29-1
2
ASS.R.*
TRBV7-3
YAML Specification:
my_mr_encoding: MatchedRegex: motif_filepath: path/to/file.txt match_v_genes: True reads: unique
MatchedSequences
Encodes the dataset based on the matches between a RepertoireDataset and a reference sequence dataset.
This encoding can be used in combination with the Matches report.
When sum_matches and normalize are set to True, this encoder behaves as described in: Yao, Y. et al. ‘T cell receptor repertoire as a potential diagnostic marker for celiac disease’. Clinical Immunology Volume 222 (January 2021): 108621. doi.org/10.1016/j.clim.2020.108621
Arguments:
reference (dict): A dictionary describing the reference dataset file. Import should be specified the same way as regular dataset import. It is only allowed to import a sequence dataset here (i.e., is_repertoire and paired are False by default, and are not allowed to be set to True).
max_edit_distance (int): The maximum edit distance between a target sequence (from the repertoire) and the reference sequence.
reads (
ReadsType): Reads type signify whether the counts of the sequences in the repertoire will be taken into account. IfUNIQUE, only unique sequences (clonotypes) are counted, and ifALL, the sequence ‘count’ value is summed when determining the number of matches. The default value for reads is all.sum_matches (bool): When sum_matches is False, the resulting encoded data matrix contains multiple columns with the number of matches per reference sequence. When sum_matches is true, all columns are summed together, meaning that there is only one aggregated sum of matches per repertoire in the encoded data. To use this encoder in combination with the Matches report, sum_matches must be set to False. When sum_matches is set to True, this encoder behaves as described by Yao, Y. et al. By default, sum_matches is False.
normalize (bool): If True, the sequence matches are divided by the total number of unique sequences in the repertoire (when reads = unique) or the total number of reads in the repertoire (when reads = all).
YAML Specification:
my_ms_encoding: MatchedSequences: reference: path: path/to/file.txt format: VDJDB max_edit_distance: 1
OneHot
One-hot encoding for repertoires, sequences or receptors. In one-hot encoding, each alphabet character (amino acid or nucleotide) is replaced by a sparse vector with one 1 and the rest zeroes. The position of the 1 represents the alphabet character.
Arguments:
use_positional_info (bool): whether to include features representing the positional information. If True, three additional feature vectors will be added, representing the sequence start, sequence middle and sequence end. The values in these features are scaled between 0 and 1. A graphical representation of the values of these vectors is given below.
Value of sequence start: Value of sequence middle: Value of sequence end: 1 \ 1 /‾‾‾‾‾‾‾‾‾‾‾‾‾‾‾‾‾\ 1 / \ / \ / \ / \ / 0 \_____________________ 0 / \ 0 _____________________/ <----sequence length----> <----sequence length----> <----sequence length---->distance_to_seq_middle (int): only applies when use_positional_info is True. This is the distance from the edge of the CDR3 sequence (IMGT positions 105 and 117) to the portion of the sequence that is considered ‘middle’. For example: if distance_to_seq_middle is 6 (default), all IMGT positions in the interval [111, 112) receive positional value 1. When using nucleotide sequences: note that the distance is measured in (amino acid) IMGT positions. If the complete sequence length is smaller than 2 * distance_to_seq_middle, the maximum value of the ‘start’ and ‘end’ vectors will not reach 0, and the maximum value of the ‘middle’ vector will not reach 1. A graphical representation of the positional vectors with a too short sequence is given below:
Value of sequence start Value of sequence middle Value of sequence end: with very short sequence: with very short sequence: with very short sequence: 1 \ 1 1 / \ / \ /\ / 0 0 / \ 0 <-> <--> <->flatten (bool): whether to flatten the final onehot matrix to a 2-dimensional matrix [examples, other_dims_combined] This must be set to True when using onehot encoding in combination with scikit-learn ML methods (inheriting
SklearnMethod), such as LogisticRegression, SVM, SVC, RandomForestClassifier and KNN.sequence_type: whether to use nucleotide or amino acid sequence for encoding. Valid values are ‘nucleotide’ and ‘amino_acid’.
YAML specification:
one_hot_vanilla: OneHot: use_positional_info: False flatten: False sequence_type: amino_acid one_hot_positional: OneHot: use_positional_info: True distance_to_seq_middle: 3 flatten: False sequence_type: nucleotide
SequenceAbundance
This encoder represents the repertoires as vectors where:
the first element corresponds to the number of label-associated clonotypes
the second element is the total number of unique clonotypes
To determine what clonotypes (with features defined by comparison_attributes) are label-associated based on a statistical test. The statistical test used is Fisher’s exact test (one-sided).
Reference: Emerson, Ryan O. et al. ‘Immunosequencing Identifies Signatures of Cytomegalovirus Exposure History and HLA-Mediated Effects on the T Cell Repertoire’. Nature Genetics 49, no. 5 (May 2017): 659–65. doi.org/10.1038/ng.3822.
Note: to use this encoder, it is necessary to explicitly define the positive class for the label when defining the label in the instruction. With positive class defined, it can then be determined which sequences are indicative of the positive class. For full example of using this encoder, see Reproduction of the CMV status predictions study.
Arguments:
comparison_attributes (list): The attributes to be considered to group receptors into clonotypes. Only the fields specified in comparison_attributes will be considered, all other fields are ignored. Valid values are sequence_aas, sequences, v_genes, j_genes, v_subgroups, j_subgroups, v_alleles, j_alleles, chains, counts, region_types, frame_types, sequence_identifiers, cell_ids.
p_value_threshold (float): The p value threshold to be used by the statistical test.
sequence_batch_size (int): The number of sequences in a batch when comparing sequences across repertoires, typically 100s of thousands. This does not affect the results of the encoding, only the speed.
repertoire_batch_size (int): How many repertoires will be loaded at once. This does not affect the result of the encoding, only the speed. This value is a trade-off between the number of repertoires that can fit the RAM at the time and loading time from disk.
YAML specification:
my_sa_encoding: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes - chains - region_types p_value_threshold: 0.05 sequence_batch_size: 100000 repertoire_batch_size: 32
TCRdist
Encodes the given ReceptorDataset as a distance matrix between all receptors, where the distance is computed using TCRdist from the paper: Dash P, Fiore-Gartland AJ, Hertz T, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017; 547(7661):89-93. doi:10.1038/nature22383.
For the implementation, TCRdist3 library was used (source code available here).
Arguments:
cores (int): number of processes to use for the computation
YAML specification:
my_tcr_dist_enc: # user-defined name TCRdist: cores: 4
Word2Vec
Word2VecEncoder learns the vector representations of k-mers based on the context (receptor sequence). It works for sequence and repertoire datasets. Similar idea was discussed in: Ostrovsky-Berman, M., Frankel, B., Polak, P. & Yaari, G. Immune2vec: Embedding B/T Cell Receptor Sequences in ℝN Using Natural Language Processing. Frontiers in Immunology 12, (2021).
This encoder relies on gensim’s implementation of Word2Vec and KmerHelper for k-mer extraction. Currently it works on amino acid level.
Arguments:
vector_size (int): The size of the vector to be learnt.
model_type (
ModelType): The context which will be used to infer the representation of the sequence. IfSEQUENCEis used, the context of a k-mer is defined by the sequence it occurs in (e.g. if the sequence is CASTTY and k-mer is AST, then its context consists of k-mers CAS, STT, TTY) IfKMER_PAIRis used, the context for the k-mer is defined as all the k-mers that within one edit distance (e.g. for k-mer CAS, the context includes CAA, CAC, CAD etc.). Valid values are SEQUENCE, KMER_PAIR.k (int): The length of the k-mers used for the encoding.
epochs (int): for how many epochs to train the word2vec model for a given set of sentences (corresponding to epochs parameter in gensim package)
window (int): max distance between two k-mers in a sequence (same as window parameter in gensim’s word2vec)
YAML specification:
encodings: my_w2v: Word2Vec: vector_size: 16 k: 3 model_type: SEQUENCE epochs: 100 window: 8
Reports
Data reports
Data reports show some type of features or statistics about a given dataset.
When running the TrainMLModel instruction, data reports can be specified under the key ‘data_reports’, to run the report on the whole dataset, or inside the ‘selection’ or ‘assessment’ specification under the keys ‘reports/data’ (current cross-validation split) or ‘reports/data_splits’ (train/test sub-splits).
Alternatively, when running the ExploratoryAnalysis instruction, data reports can be specified under ‘reports’.
When using the reports with instructions such as ExploratoryAnalysis or TrainMLModel, the arguments defined below are set at runtime by the instruction. Concrete classes inheriting DataReport may include additional parameters that will be set by the user in the form of input arguments.
Arguments:
dataset (Dataset): a dataset object (can be repertoire, receptor or sequence dataset, depending on the specific report)
result_path (Path): location where the results (plots, tables, etc.) will be stored
name (str): user-defined name of the report used in the HTML overview automatically generated by the platform
number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
CytoscapeNetworkExporter
This report exports the Receptor sequences to .sif format, such that they can directly be imported as a network in Cytoscape, to visualize chain sharing between the different receptors in a dataset (for example, for TCRs: how often one alpha chain is shared with multiple beta chains, and vice versa).
The Receptor sequences can be provided as a ReceptorDataset, or a RepertoireDataset (containing paired sequence information). In the latter case, one .sif file is exported per Repertoire.
YAML specification:
my_cyto_export: CytoscapeNetworkExporter
GLIPH2Exporter
Report which exports the receptor data to GLIPH2 format so that it can be directly used in GLIPH2 tool. Currently, the report accepts only receptor datasets.
GLIPH2 publication: Huang H, Wang C, Rubelt F, Scriba TJ, Davis MM. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nature Biotechnology. Published online April 27, 2020:1-9. doi:10.1038/s41587-020-0505-4
Arguments:
condition (str): name of the parameter present in the receptor metadata in the dataset; condition can be anything which can be processed in GLIPH2, such as tissue type or treatment.
YAML specification:
my_gliph2_exporter: # user-defined name GLIPH2Exporter: condition: epitope # for instance, epitope parameter is present in receptors' metadata with values such as "MtbLys" for Mycobacterium tuberculosis (as shown in the original paper).
ReceptorDatasetOverview
This report plots the length distribution per chain for a receptor (paired-chain) dataset.
Arguments:
batch_size (int): how many receptors to load at once; 50 000 by default
YAML specification:
reports: my_receptor_overview_report: ReceptorDatasetOverview
SequenceLengthDistribution
Generates a histogram of the lengths of the sequences in a RepertoireDataset.
YAML specification:
my_sld_report: SequenceLengthDistribution
SimpleDatasetOverview
Generates a simple text-based overview of the properties of any dataset, including the dataset name, size, and metadata labels.
YAML specification:
reports: my_overview: SimpleDatasetOverview
Encoding reports
Encoding reports show some type of features or statistics about an encoded dataset, or may in some cases export relevant sequences or tables.
When running the TrainMLModel instruction, encoding reports can be specified inside the ‘selection’ or ‘assessment’ specification under the key ‘reports:encoding’. Alternatively, when running the ExploratoryAnalysis instruction, encoding reports can be specified under ‘reports’.
When using the reports with instructions such as ExploratoryAnalysis or TrainMLModel, the arguments defined below are set at runtime by the instruction. Concrete classes inheriting EncodingReport may include additional parameters that will be set by the user in the form of input arguments.
Arguments:
dataset (Dataset): an encoded dataset where encoded_data attribute is set to an instance of EncodedData object
result_path (Path): path where the results will be stored (plots, tables, etc.)
name (str): user-defined name of the report that will be shown in the HTML overview later
number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
DesignMatrixExporter
Exports the design matrix and related information of a given encoded Dataset to csv files. If the encoded data has more than 2 dimensions (such as when using the OneHot encoder with option Flatten=False), the data are then exported to different formats to facilitate their import with external software.
Arguments:
file_format (str): the format and extension of the file to store the design matrix. The supported formats are: npy, csv, hdf5, npy.zip, csv.zip or hdf5.zip.
YAML specification:
my_dme_report: DesignMatrixExporter: file_format: csv
FeatureComparison
Compares the feature values in a given encoded data matrix across two values for a metadata label. These labels are specified in the metadata file for repertoire datasets, or as metadata columns for sequence and receptor datasets. Can be used in combination with any encoding and dataset type. This report produces a scatterplot, where each point represents one feature, and the values on the x and y axes are the average feature values across two subsets of the data. For example, when KmerFrequency encoder is used, and the comparison_label is used to represent a disease (true/false), then the features are the k-mers (AAA, AAC, etc..) and their x and y position in the scatterplot is determined by their frequency in the subset of the data where disease=true and disease=false.
Optional metadata labels can be specified to divide the scatterplot into groups based on color, row facets or column facets.
Alternatively, when the feature values are of interest without comparing them between labelled subgroups of the data, please use FeatureValueBarplot or FeatureDistribution instead.
Arguments:
comparison_label (str): Mandatory label. This label is used to split the encoded data matrix and define the x and y axes of the plot. This label is only allowed to have 2 classes (for example: sick and healthy, binding and non-binding).
color_grouping_label (str): Optional label that is used to color the points in the scatterplot. This can not be the same as comparison_label.
row_grouping_label (str): Optional label that is used to group scatterplots into different row facets. This can not be the same as comparison_label.
column_grouping_label (str): Optional label that is used to group scatterplots into different column facets. This can not be the same as comparison_label.
show_error_bar (bool): Whether to show the error bar (standard deviation) for the points, both in the x and y dimension.
log_scale (bool): Whether to plot the x and y axes in log scale (log_scale = True) or continuous scale (log_scale = False). By default, log_scale is False.
keep_fraction (float): The total number of features may be very large and only the features differing significantly across comparison labels may be of interest. When the keep_fraction parameter is set below 1, only the fraction of features that differs the most across comparison labels is kept for plotting (note that the produced .csv file still contains all data). By default, keep_fraction is 1, meaning that all features are plotted.
YAML specification:
my_comparison_report: FeatureComparison: # compare the different classes defined in the label disease comparison_label: disease
FeatureDistribution
Plots a boxplot for each feature in the encoded data matrix. Can be used in combination with any encoding and dataset type. Each boxplot represents a feature and shows the distribution of values for that feature. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
Two modes can be used: in the ‘normal’ mode there are normal boxplots corresponding to each column of the encoded dataset matrix; in the ‘sparse’ mode all zero cells are eliminated before passing the data to the boxplots. If mode is set to ‘auto’, then it will automatically set to ‘sparse’ if the density of the matrix is below 0.01
Optional metadata labels can be specified to divide the boxplots into groups based on color, row facets or column facets. These labels are specified in the metadata file for repertoire datasets, or as metadata columns for sequence and receptor datasets.
Alternatively, when only the mean feature values are of interest (as opposed to showing the complete distribution, as done here), please consider using FeatureValueBarplot instead. When comparing the feature values between two subsets of the data, please use FeatureComparison.
Arguments:
color_grouping_label (str): The label that is used to color each bar, at each level of the grouping_label.
row_grouping_label (str): The label that is used to group bars into different row facets.
column_grouping_label (str): The label that is used to group bars into different column facets.
mode (str): either ‘normal’, ‘sparse’ or ‘auto’ (default)
x_title (str): x-axis label
y_title (str): y-axis label
YAML specification:
my_fdistr_report: FeatureDistribution: mode: sparse
FeatureValueBarplot
Plots a barplot of the feature values in a given encoded data matrix, averaged across examples. Can be used in combination with any encoding and dataset type. Each bar in the barplot represents the mean value of a given feature, and along the x-axis are the different features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
Optional metadata labels can be specified to divide the barplot into groups based on color, row facets or column facets. In this case, the average feature values in each group are plotted. These labels are specified in the metadata file for repertoire datasets, or as metadata columns for sequence and receptor datasets.
Alternatively, when the distribution of feature values is of interest (as opposed to showing only the mean, as done here), please consider using FeatureDistribution instead. When comparing the feature values between two subsets of the data, please use FeatureComparison.
Arguments:
color_grouping_label (str): The label that is used to color each bar, at each level of the grouping_label.
row_grouping_label (str): The label that is used to group bars into different row facets.
column_grouping_label (str): The label that is used to group bars into different column facets.
show_error_bar (bool): Whether to show the error bar (standard deviation) for the bars.
x_title (str): x-axis label
y_title (str): y-axis label
YAML specification:
my_fvb_report: FeatureValueBarplot: # timepoint, disease_status and age_group are metadata labels column_grouping_label: timepoint row_grouping_label: disease_status color_grouping_label: age_group
Matches
Reports the number of matches that were found when using one of the following encoders:
MatchedSequences encoder
MatchedReceptors encoder
MatchedRegex encoder
Report results are:
A table containing all matches, where the rows correspond to the Repertoires, and the columns correspond to the objects to match (regular expressions or receptor sequences).
The repertoire sizes (read frequencies and the number of unique sequences per repertoire), for each of the chains. This can be used to calculate the percentage of matched sequences in a repertoire.
When using MatchedSequences encoder or MatchedReceptors encoder, tables describing the chains and receptors (ids, chains, V and J genes and sequences).
When using MatchedReceptors encoder or using MatchedRegex encoder with chain pairs, tables describing the paired matches (where a match was found in both chains) per repertoire.
YAML Specification:
my_match_report: Matches
RelevantSequenceExporter
Exports the sequences that are extracted as label-associated using the SequenceAbundance encoder in AIRR-compliant format.
Arguments: there are no arguments for this report.
YAML specification:
my_relevant_sequences: RelevantSequenceExporter
ML model reports
ML model reports show some type of features or statistics about one trained ML model.
In the TrainMLModel instruction, ML model reports can be specified inside the ‘selection’ or ‘assessment’ specification under the key ‘reports:models’.
When using the reports with TrainMLModel instruction, the arguments defined below are set at runtime by the instruction. Concrete classes inheriting MLReport may include additional parameters that will be set by the user in the form of input arguments.
Arguments:
train_dataset (Dataset): a dataset object (repertoire, receptor or sequence dataset) with encoded_data attribute set to an EncodedData object that was used for training the ML method
test_dataset (Dataset): same as train_dataset, except it is not used for training and then maybe be used for testing the method
method (MLMethod): a trained instance of a concrete subclass of MLMethod object
result_path (Path): location where the report results will be stored
hp_setting (HPSetting): a HPSetting object describing the ML method, encoding and preprocessing used
label (Label): the label for which the model was trained
name (str): user-defined name of the report used in the HTML overview automatically generated by the platform
number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
Coefficients
A report that plots the coefficients for a given ML method in a barplot. Can be used for LogisticRegression, SVM, SVC, and RandomForestClassifier. In the case of RandomForest, the feature importances will be plotted.
When used in TrainMLModel instruction, the report can be specified under ‘models’, both on the selection and assessment levels.
Which coefficients should be plotted (for example: only nonzero, above a certain threshold, …) can be specified. Multiple options can be specified simultaneously. By default the 25 largest coefficients are plotted. The full set of coefficients will also be exported as a csv file.
Arguments:
coefs_to_plot (list): A list specifying which coefficients should be plotted. Valid values are: ALL, NONZERO, CUTOFF, N_LARGEST.
cutoff (list): If ‘cutoff’ is specified under ‘coefs_to_plot’, the cutoff values can be specified here. The coefficients which have an absolute value equal to or greater than the cutoff will be plotted.
n_largest (list): If ‘n_largest’ is specified under ‘coefs_to_plot’, the values for n can be specified here. These should be integer values. The n largest coefficients are determined based on their absolute values.
YAML specification:
my_coef_report: Coefficients: coefs_to_plot: - all - nonzero - cutoff - n_largest cutoff: - 0.1 - 0.01 n_largest: - 5 - 10
ConfounderAnalysis
A report that plots the numbers of false positives and false negatives with respect to each value of the metadata features specified by the user. This allows checking whether a given machine learning model makes more misclassifications for some values of a metadata feature than for the others.
Arguments:
metadata_labels (list): A list of the metadata features to use as a basis for the calculations
YAML specification:
my_confounder_report: ConfounderAnalysis: metadata_labels: - age - sex
DeepRCMotifDiscovery
This report plots the contributions of (i) input sequences and (ii) kernels to trained DeepRC model with respect to the test dataset. Contributions are computed using integrated gradients (IG). This report produces two figures:
inputs_integrated_gradients: Shows the contributions of the characters within the input sequences (test dataset) that was most important for immune status prediction of the repertoire. IG is only applied to sequences of positive class repertoires.
kernel_integrated_gradients: Shows the 1D CNN kernels with the highest contribution over all positions and amino acids.
For both inputs and kernels: Larger characters in the extracted motifs indicate higher contribution, with blue indicating positive contribution and red indicating negative contribution towards the prediction of the immune status. For kernels only: contributions to positional encoding are indicated by < (beginning of sequence), ∧ (center of sequence), and > (end of sequence).
See DeepRCMotifDiscovery for repertoire classification for a usage example.
Reference: Michael Widrich, Bernhard Schäfl, Milena Pavlović, Geir Kjetil Sandve, Sepp Hochreiter, Victor Greiff, Günter Klambauer ‘DeepRC: Immune repertoire classification with attention-based deep massive multiple instance learning’. bioRxiv preprint doi: https://doi.org/10.1101/2020.04.12.03815
Arguments:
n_steps (int): Number of IG steps (more steps -> better path integral -> finer contribution values). 50 is usually good enough.
threshold (float): Only applies to the plotting of kernels. Contributions are normalized to range [0, 1], and only kernels with normalized contributions above threshold are plotted.
YAML specification:
my_deeprc_report: DeepRCMotifDiscovery: threshold: 0.5 n_steps: 50
KernelSequenceLogo
A report that plots kernels of a CNN model as sequence logos. It works only with trained ReceptorCNN models which has kernels already normalized to represent information gain matrices. Additionally, it also plots the weights in the final fully-connected layer of the network associated with kernel outputs. For more information on how the model works, see ReceptorCNN.
The kernels are visualized using Logomaker. Original publication: Tareen A, Kinney JB. Logomaker: beautiful sequence logos in Python. Bioinformatics. 2020; 36(7):2272-2274. doi:10.1093/bioinformatics/btz921.
Arguments: this report does not take any arguments as input.
YAML specification:
my_kernel_seq_logo: KernelSequenceLogo
MotifSeedRecovery
This report can be used to show how well implanted motifs (for example, through the Simulation instruction) can be recovered by various machine learning methods using the k-mer encoding. This report creates a boxplot, where the x axis (box grouping) represents the maximum possible overlap between an implanted motif seed and a kmer feature (measured in number of positions), and the y axis shows the coefficient size of the respective kmer feature. If the machine learning method has learned the implanted motif seeds, the coefficient size is expected to be largest for the kmer features with high overlap to the motif seeds.
Note that to use this report, the following criteria must be met:
KmerFrequencyEncoder must be used.
One of the following classifiers must be used: RandomForestClassifier, LogisticRegression, SVM, SVC
For each label, the implanted motif seeds relevant to that label must be specified
To find the overlap score between kmer features and implanted motif seeds, the two sequences are compared in a sliding window approach, and the maximum overlap is calculated.
Overlap scores between kmer features and implanted motifs are calculated differently based on the Hamming distance that was allowed during implanting.
Without hamming distance: Seed: AAA -> score = 3 Feature: xAAAx ^^^ Seed: AAA -> score = 0 Feature: xAAxx With hamming distance: Seed: AAA -> score = 3 Feature: xAAAx ^^^ Seed: AAA -> score = 2 Feature: xAAxx ^^ Furthermore, gap positions in the motif seed are ignored: Seed: A/AA -> score = 3 Feature: xAxAAx ^/^^See Recovering simulated immune signals for more details and an example plot.
Arguments:
implanted_motifs_per_label (dict): a nested dictionary that specifies the motif seeds that were implanted in the given dataset. The first level of keys in this dictionary represents the different labels. In the inner dictionary there should be two keys: “seeds” and “hamming_distance”:
seeds: a list of motif seeds. The seeds may contain gaps, specified by a ‘/’ symbol.
hamming_distance: A boolean value that specifies whether hamming distance was allowed when implanting the motif seeds for a given label. Note that this applies to all seeds for this label.
gap_sizes: a list of all the possible gap sizes that were used when implanting a gapped motif seed. When no gapped seeds are used, this value has no effect.
YAML specification:
my_motif_report: MotifSeedRecovery: implanted_motifs_per_label: CD: seeds: - AA/A - AAA hamming_distance: False gap_sizes: - 0 - 1 - 2 T1D: seeds: - CC/C - CCC hamming_distance: True gap_sizes: - 2
ROCCurve
A report that plots the ROC curve for a binary classifier.
YAML specification:
reports: my_roc_report: ROCCurve
SequenceAssociationLikelihood
Plots the beta distribution used as a prior for class assignment in ProbabilisticBinaryClassifier. The distribution plotted shows the probability that a sequence is associated with a given class for a label.
Attributes: the report does not take in any arguments.
YAML specification:
my_sequence_assoc_report: SequenceAssociationLikelihood
TCRdistMotifDiscovery
The report for discovering motifs in paired immune receptor data of given specificity based on TCRdist3. The receptors are hierarchically clustered based on the tcrdist distance and then motifs are discovered for each cluster. The report outputs logo plots for the motifs along with the raw data used for plotting in csv format.
For the implementation, TCRdist3 library was used (source code available here). More details on the functionality used for this report are available here.
Original publications:
Dash P, Fiore-Gartland AJ, Hertz T, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017; 547(7661):89-93. doi:10.1038/nature22383
Mayer-Blackwell K, Schattgen S, Cohen-Lavi L, et al. TCR meta-clonotypes for biomarker discovery with tcrdist3: quantification of public, HLA-restricted TCR biomarkers of SARS-CoV-2 infection. bioRxiv. Published online December 26, 2020:2020.12.24.424260. doi:10.1101/2020.12.24.424260
Arguments:
positive_class_name (str): the class value (e.g., epitope) used to select only the receptors that are specific to the given epitope so that only those sequences are used to infer motifs; the reference receptors as required by TCRdist will be the ones from the dataset that have different or no epitope specified in their metadata; if the labels are available only on the epitope level (e.g., label is “AVFDRKSDAK” and classes are True and False), then here it should be specified that only the receptors with value “True” for label “AVFDRKSDAK” should be used; there is no default value for this argument
cores (int): number of processes to use for the computation of the distance and motifs
min_cluster_size (int): the minimum size of the cluster to discover the motifs for
use_reference_sequences (bool): when showing motifs, this parameter defines if reference sequences should be provided as well as a background
YAML specification:
my_tcr_dist_report: # user-defined name TCRdistMotifDiscovery: positive_class_name: True # class name, could also be epitope name, depending on how it's defined in the dataset cores: 4 min_cluster_size: 30 use_reference_sequences: False
TrainingPerformance
A report that plots the evaluation metrics for the performance given machine learning model and training dataset. The available metrics are accuracy, balanced_accuracy, confusion_matrix, f1_micro, f1_macro, f1_weighted, precision, recall, auc and log_loss (see
immuneML.environment.Metric.Metric).Arguments:
metrics (list): A list of metrics used to evaluate training performance. See
immuneML.environment.Metric.Metricfor available options.YAML specification:
my_performance_report: TrainingPerformance: metrics: - accuracy - balanced_accuracy - confusion_matrix - f1_micro - f1_macro - f1_weighted - precision - recall - auc - log_loss
Train ML model reports
Train ML model reports plot general statistics or export data of multiple models simultaneously when running the TrainMLModel instruction.
In the TrainMLModel instruction, train ML model reports can be specified under ‘reports’.
When using the reports with TrainMLModel instruction, the arguments defined below are set at runtime by the instruction. Concrete classes inheriting TrainMLModelReport may include additional parameters that will be set by the user in the form of input arguments.
Arguments:
name (str): user-defined name of the report used in the HTML overview automatically generated by the platform
state (TrainMLModelState): a state object that includes all the information, trained models, encodings and datasets from the nested cross-validation procedure used to train the optimal model.
result_path (Path): location where the report results will be stored
number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
CVFeaturePerformance
This report plots the average training vs test performance w.r.t. given encoding parameter which is explicitly set in the feature attribute. It can be used only in combination with TrainMLModel instruction and can be only specified under ‘reports’
- Arguments:
feature: name of the encoder parameter w.r.t. which the performance across training and test will be shown. Possible values depend on the encoder on which it is used.
is_feature_axis_categorical (bool): if the x-axis of the plot where features are shown should be categorical; alternatively it is automatically determined based on the feature values
YAML specification:
report1: CVFeaturePerformance: feature: p_value_threshold # parameter value of SequenceAbundance encoder is_feature_axis_categorical: True # show x-axis as categorical
DiseaseAssociatedSequenceCVOverlap
DiseaseAssociatedSequenceCVOverlap report makes one heatmap per label showing the overlap of disease-associated sequences produced by the SequenceAbundance encoder between folds of cross-validation (either inner or outer loop of the nested CV). The overlap is computed by the following equation:
\[overlap(X,Y) = \frac{|X \cap Y|}{min(|X|, |Y|)} x 100\]For details, see Greiff V, Menzel U, Miho E, et al. Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Reports. 2017;19(7):1467-1478. doi:10.1016/j.celrep.2017.04.054.
Arguments:
compare_in_selection (bool): whether to compute the overlap over the inner loop of the nested CV - the sequence overlap is shown across CV folds for the model chosen as optimal within that selection
compare_in_assessment (bool): whether to compute the overlap over the optimal models in the outer loop of the nested CV
YAML specification:
reports: # the report is defined with all other reports under definitions/reports my_overlap_report: DiseaseAssociatedSequenceCVOverlap # report has no parameters
MLSettingsPerformance
Report for TrainMLModel instruction: plots the performance for each of the setting combinations as defined under ‘settings’ in the assessment (outer validation) loop.
The performances are grouped by label (horizontal panels) encoding (vertical panels) and ML method (bar color). When multiple data splits are used, the average performance over the data splits is shown with an error bar representing the standard deviation.
This report can be used only with TrainMLModel instruction under ‘reports’.
Arguments:
single_axis_labels (bool): whether to use single axis labels. Note that using single axis labels makes the figure unsuited for rescaling, as the label position is given in a fixed distance from the axis. By default, single_axis_labels is False, resulting in standard plotly axis labels.
x_label_position (float): if single_axis_labels is True, this should be an integer specifying the x axis label position relative to the x axis. The default value for label_position is -0.1.
y_label_position (float): same as x_label_position, but for the y axis.
YAML specification:
my_hp_report: MLSettingsPerformance
MLSubseqPerformance
Report for TrainMLModel: Similar to
MLSettingsPerformance, this report plots the performance of certain combinations of encodings and ML methods.Similarly to MLSettingsPerformance, the performances are grouped by label (horizontal panels). However, the bar color is determined by the ml method class (thus several ML methods with different parameters may be grouped together) and the vertical panel grouping is determined by the subsequence size used for motif recovery. This subsequence size is either the k-mer size or the kernel size (DeepRC).
This report can only be used to plot the results for setting combinations using k-mer encoding with continuous k-mers (in combination with any ML method), or DeepRC encoding + ml method.
This report can only be used with TrainMLModel instruction under ‘reports’.
YAML specification:
my_hp_report: MLSubseqPerformance
ROCCurveSummary
This report plots ROC curves for all trained ML settings ([preprocessing], encoding, ML model) in the outer loop of cross-validation in the TrainMLModel instruction. If there are multiple splits in the outer loop, this report will make one plot per split. This report is defined only for binary classification. If there are multiple labels defined in the instruction, each label has to have two classes to be included in this report.
Arguments: there are no arguments for this report.
YAML specification:
- reports:
my_roc_summary_report: ROCCurveSummary
ReferenceSequenceOverlap
The ReferenceSequenceOverlap report compares a list of disease-associated sequences produced by the SequenceAbundance encoder to a list of reference receptor sequences. It outputs a Venn diagram and a list of receptor sequences found both in the encoder and reference.
The report compares the sequences by their sequence content and the additional comparison_attributes (such as V or J gene), as specified by the user.
Arguments:
reference_path (str): path to the reference file in csv format which contains one entry per row and has columns that correspond to the attributes listed under comparison_attributes argument
comparison_attributes (list): list of attributes to use for comparison; all of them have to be present in the reference file where they should be the names of the columns
label (str): name of the label for which the reference sequences should be compared to the model; if none, it takes the one label from the instruction; if it is none and multiple labels were specified for the instruction, the report will not be generated
YAML specification:
reports: # the report is defined with all other reports under definitions/reports my_reference_overlap_report: ReferenceSequenceOverlap: reference_path: reference.csv # a reference file with columns listed under comparison_attributes comparison_attributes: - sequence_aas - v_genes - j_genes
Multi dataset reports
Multi dataset reports are special reports that can be specified when running immuneML with the
MultiDatasetBenchmarkTool.When running the
MultiDatasetBenchmarkTool, multi dataset reports can be specified under ‘benchmark_reports’.When using the reports with MultiDatasetBenchmarkTool, the arguments defined below are set at runtime by the instruction. Concrete classes inheriting MultiDatasetReport may include additional parameters that will be set by the user in the form of input arguments.
Arguments:
name (str): user-defined name of the report used in the HTML overview automatically generated by the platform
result_path (Path): location where the report results will be stored
instruction_states (list): a list of states for each instruction that was run as a part of the tool, e.g., TrainMLModelState objects
number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
DiseaseAssociatedSequenceOverlap
DiseaseAssociatedSequenceOverlap report makes a heatmap showing the overlap of disease-associated sequences produced by SequenceAbundance encoders between multiple datasets of different sizes (different number of repertoires per dataset).
This plot can be used only with MultiDatasetBenchmarkTool
The overlap is computed by the following equation:
\[overlap(X,Y) = \frac{|X \cap Y|}{min(|X|, |Y|)} x 100\]For details, see Greiff V, Menzel U, Miho E, et al. Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Reports. 2017;19(7):1467-1478. doi:10.1016/j.celrep.2017.04.054.
YAML specification:
reports: # the report is defined with all other reports under definitions/reports my_overlap_report: DiseaseAssociatedSequenceOverlap # report has no parameters
PerformanceOverview
PerformanceOverview report creates an ROC plot and precision-recall plot for optimal trained models on multiple datasets. The labels on the plots are the names of the datasets, so it might be good to have user-friendly names when defining datasets that are still a combination of letters, numbers and the underscore sign.
This report can be used only with MultiDatasetBenchmarkTool as it will plot ROC and PR curve for trained models across datasets. Also, it requires the task to be immune repertoire classification and cannot be used for receptor or sequence classification. Furthermore, it uses predictions on the test dataset to assess the performance and plot the curves. If the parameter refit_optimal_model is set to True, all data will be used to fit the optimal model, so there will not be a test dataset which can be used to assess performance and the report will not be generated.
If datasets have the same number of examples, the baseline PR curve will be plotted as described in this publication: Saito T, Rehmsmeier M. The Precision-Recall Plot Is More Informative than the ROC Plot When Evaluating Binary Classifiers on Imbalanced Datasets. PLOS ONE. 2015;10(3):e0118432. doi:10.1371/journal.pone.0118432
If the datasets have different number of examples, the baseline PR curve will not be plotted.
YAML specification:
reports: my_performance_report: PerformanceOverview
ML methods
When choosing which ML method(s) are most suitable for your use-case, please consider the following table. The table describes which of the ML methods can be used for binary classification (two classes per label), and which can be used for multi-class classification. Note that all classifiers can automatically be used for multi-label classification in immuneML. Furthermore, it describes what type of dataset the classifier can be applied to, and whether a third level of nested cross-validation can be used for the selection of model parameters (scikit-learn classifiers).
ML method |
binary classification |
multi-class classification |
sequence dataset |
receptor dataset |
repertoire dataset |
model selection CV |
|---|---|---|---|---|---|---|
AtchleyKmerMILClassifier |
✓ |
✗ |
✗ |
✗ |
✓ |
✗ |
DeepRC |
✓ |
✗ |
✗ |
✗ |
✓ |
✗ |
KNN |
✓ |
✓ |
✓ |
✓ |
✓ |
✓ |
LogisticRegression |
✓ |
✓ |
✓ |
✓ |
✓ |
✓ |
ProbabalisticBinaryClassifier |
✓ |
✗ |
✗ |
✗ |
✓ |
✗ |
RandomForestClassifier |
✓ |
✓ |
✓ |
✓ |
✓ |
✓ |
ReceptorCNN |
✓ |
✗ |
✗ |
✓ |
✗ |
✗ |
SVM |
✓ |
✓ |
✓ |
✓ |
✓ |
✓ |
TCRdistClassifier |
✓ |
✓ |
✓ |
✓ |
✓ |
✗ |
AtchleyKmerMILClassifier
A binary Repertoire classifier which uses the data encoded by AtchleyKmer encoder to predict the repertoire label.
The original publication: Ostmeyer J, Christley S, Toby IT, Cowell LG. Biophysicochemical motifs in T cell receptor sequences distinguish repertoires from tumor-infiltrating lymphocytes and adjacent healthy tissue. Cancer Res. Published online January 1, 2019:canres.2292.2018. doi:10.1158/0008-5472.CAN-18-2292 .
Arguments:
iteration_count (int): max number of training iterations
threshold (float): loss threshold at which to stop training if reached
evaluate_at (int): log model performance every ‘evaluate_at’ iterations and store the model every ‘evaluate_at’ iterations if early stopping is used
use_early_stopping (bool): whether to use early stopping
learning_rate (float): learning rate for stochastic gradient descent
random_seed (int): random seed used
zero_abundance_weight_init (bool): whether to use 0 as initial weight for abundance term (if not, a random value is sampled from normal distribution with mean 0 and variance 1 / total_number_of_features
number_of_threads: number of threads to be used for training
YAML specification:
my_kmer_mil_classifier: AtchleyKmerMILClassifier: iteration_count: 100 evaluate_at: 15 use_early_stopping: False learning_rate: 0.01 random_seed: 100 zero_abundance_weight_init: True number_of_threads: 8 threshold: 0.00001
DeepRC
This classifier uses the DeepRC method for repertoire classification. The DeepRC ML method should be used in combination with the DeepRC encoder. Also consider using the DeepRCMotifDiscovery report for interpretability.
Notes:
DeepRC uses PyTorch functionalities that depend on GPU. Therefore, DeepRC does not work on a CPU.
This wrapper around DeepRC currently only supports binary classification.
Reference: Michael Widrich, Bernhard Schäfl, Milena Pavlović, Geir Kjetil Sandve, Sepp Hochreiter, Victor Greiff, Günter Klambauer ‘DeepRC: Immune repertoire classification with attention-based deep massive multiple instance learning’. bioRxiv preprint doi: https://doi.org/10.1101/2020.04.12.038158
Arguments:
validation_part (float): the part of the data that will be used for validation, the rest will be used for training.
add_positional_information (bool): whether positional information should be included in the input features.
kernel_size (int): the size of the 1D-CNN kernels.
n_kernels (int): the number of 1D-CNN kernels in each layer.
n_additional_convs (int): Number of additional 1D-CNN layers after first layer
n_attention_network_layers (int): Number of attention layers to compute keys
n_attention_network_units (int): Number of units in each attention layer
n_output_network_units (int): Number of units in the output layer
consider_seq_counts (bool): whether the input data should be scaled by the receptor sequence counts.
sequence_reduction_fraction (float): Fraction of number of sequences to which to reduce the number of sequences per bag based on attention weights. Has to be in range [0,1].
reduction_mb_size (int): Reduction of sequences per bag is performed using minibatches of reduction_mb_size` sequences to compute the attention weights.
n_updates (int): Number of updates to train for
n_torch_threads (int): Number of parallel threads to allow PyTorch
learning_rate (float): Learning rate for adam optimizer
l1_weight_decay (float): l1 weight decay factor. l1 weight penalty will be added to loss, scaled by l1_weight_decay
l2_weight_decay (float): l2 weight decay factor. l2 weight penalty will be added to loss, scaled by l2_weight_decay
evaluate_at (int): Evaluate model on training and validation set every evaluate_at updates. This will also check for a new best model for early stopping.
sample_n_sequences (int): Optional random sub-sampling of sample_n_sequences sequences per repertoire. Number of sequences per repertoire might be smaller than sample_n_sequences if repertoire is smaller or random indices have been drawn multiple times. If None, all sequences will be loaded for each repertoire.
training_batch_size (int): Number of repertoires per minibatch during training.
n_workers (int): Number of background processes to use for converting dataset to hdf5 container and training set data loader.
pytorch_device_name (str): The name of the pytorch device to use. This name will be passed to torch.device(self.pytorch_device_name). The default value is cuda:0
YAML specification:
my_deeprc_method: DeepRC: validation_part: 0.2 add_positional_information: True kernel_size: 9
KNN
This is a wrapper of scikit-learn’s KNeighborsClassifier class. This ML method creates a distance matrix using the given encoded data. If the encoded data is already a distance matrix (for example, when using the Distance or CompAIRRDistance encoders), please use PrecomputedKNN instead.
Please see the scikit-learn documentation of KNeighborsClassifier for the parameters.
Scikit-learn models can be trained in two modes:
1. Creating a model using a given set of hyperparameters, and relying on the selection and assessment loop in the TrainMLModel instruction to select the optimal model.
2. Passing a range of different hyperparameters to KNN, and using a third layer of nested cross-validation to find the optimal hyperparameters through grid search. In this case, only the KNN model with the optimal hyperparameter settings is further used in the inner selection loop of the TrainMLModel instruction.
By default, mode 1 is used. In order to use mode 2, model_selection_cv and model_selection_n_folds must be set.
Arguments:
KNN (dict): Under this key, hyperparameters can be specified that will be passed to the scikit-learn class. Any scikit-learn hyperparameters can be specified here. In mode 1, a single value must be specified for each of the scikit-learn hyperparameters. In mode 2, it is possible to specify a range of different hyperparameters values in a list. It is also allowed to mix lists and single values in mode 2, in which case the grid search will only be done for the lists, while the single-value hyperparameters will be fixed. In addition to the scikit-learn hyperparameters, parameter show_warnings (True/False) can be specified here. This determines whether scikit-learn warnings, such as convergence warnings, should be printed. By default show_warnings is True.
model_selection_cv (bool): If any of the hyperparameters under KNN is a list and model_selection_cv is True, a grid search will be done over the given hyperparameters, using the number of folds specified in model_selection_n_folds. By default, model_selection_cv is False.
model_selection_n_folds (int): The number of folds that should be used for the cross validation grid search if model_selection_cv is True.
YAML specification:
my_knn_method: KNN: # sklearn parameters (same names as in original sklearn class) weights: uniform # always use this setting for weights n_neighbors: [5, 10, 15] # find the optimal number of neighbors # Additional parameter that determines whether to print convergence warnings show_warnings: True # if any of the parameters under KNN is a list and model_selection_cv is True, # a grid search will be done over the given parameters, using the number of folds specified in model_selection_n_folds, # and the optimal model will be selected model_selection_cv: True model_selection_n_folds: 5 # alternative way to define ML method with default values: my_default_knn: KNN
LogisticRegression
This is a wrapper of scikit-learn’s LogisticRegression class. Please see the scikit-learn documentation of LogisticRegression for the parameters.
Note: if you are interested in plotting the coefficients of the logistic regression model, consider running the Coefficients report.
Scikit-learn models can be trained in two modes:
1. Creating a model using a given set of hyperparameters, and relying on the selection and assessment loop in the TrainMLModel instruction to select the optimal model.
2. Passing a range of different hyperparameters to LogisticRegression, and using a third layer of nested cross-validation to find the optimal hyperparameters through grid search. In this case, only the LogisticRegression model with the optimal hyperparameter settings is further used in the inner selection loop of the TrainMLModel instruction.
By default, mode 1 is used. In order to use mode 2, model_selection_cv and model_selection_n_folds must be set.
Arguments:
LogisticRegression (dict): Under this key, hyperparameters can be specified that will be passed to the scikit-learn class. Any scikit-learn hyperparameters can be specified here. In mode 1, a single value must be specified for each of the scikit-learn hyperparameters. In mode 2, it is possible to specify a range of different hyperparameters values in a list. It is also allowed to mix lists and single values in mode 2, in which case the grid search will only be done for the lists, while the single-value hyperparameters will be fixed. In addition to the scikit-learn hyperparameters, parameter show_warnings (True/False) can be specified here. This determines whether scikit-learn warnings, such as convergence warnings, should be printed. By default show_warnings is True.
model_selection_cv (bool): If any of the hyperparameters under LogisticRegression is a list and model_selection_cv is True, a grid search will be done over the given hyperparameters, using the number of folds specified in model_selection_n_folds. By default, model_selection_cv is False.
model_selection_n_folds (int): The number of folds that should be used for the cross validation grid search if model_selection_cv is True.
YAML specification:
my_logistic_regression: # user-defined method name LogisticRegression: # name of the ML method # sklearn parameters (same names as in original sklearn class) penalty: l1 # always use penalty l1 C: [0.01, 0.1, 1, 10, 100] # find the optimal value for C # Additional parameter that determines whether to print convergence warnings show_warnings: True # if any of the parameters under LogisticRegression is a list and model_selection_cv is True, # a grid search will be done over the given parameters, using the number of folds specified in model_selection_n_folds, # and the optimal model will be selected model_selection_cv: True model_selection_n_folds: 5 # alternative way to define ML method with default values: my_default_logistic_regression: LogisticRegression
PrecomputedKNN
This is a wrapper of scikit-learn’s KNeighborsClassifier class. This ML method takes a pre-computed distance matrix, as created by the Distance or CompAIRRDistance encoders. If you would like to use a different encoding in combination with KNN, please use KNN instead.
Please see the scikit-learn documentation of KNeighborsClassifier for the parameters.
Scikit-learn models can be trained in two modes:
1. Creating a model using a given set of hyperparameters, and relying on the selection and assessment loop in the TrainMLModel instruction to select the optimal model.
2. Passing a range of different hyperparameters to KNN, and using a third layer of nested cross-validation to find the optimal hyperparameters through grid search. In this case, only the KNN model with the optimal hyperparameter settings is further used in the inner selection loop of the TrainMLModel instruction.
By default, mode 1 is used. In order to use mode 2, model_selection_cv and model_selection_n_folds must be set.
Arguments:
KNN (dict): Under this key, hyperparameters can be specified that will be passed to the scikit-learn class. Any scikit-learn hyperparameters can be specified here. In mode 1, a single value must be specified for each of the scikit-learn hyperparameters. In mode 2, it is possible to specify a range of different hyperparameters values in a list. It is also allowed to mix lists and single values in mode 2, in which case the grid search will only be done for the lists, while the single-value hyperparameters will be fixed. In addition to the scikit-learn hyperparameters, parameter show_warnings (True/False) can be specified here. This determines whether scikit-learn warnings, such as convergence warnings, should be printed. By default show_warnings is True.
model_selection_cv (bool): If any of the hyperparameters under KNN is a list and model_selection_cv is True, a grid search will be done over the given hyperparameters, using the number of folds specified in model_selection_n_folds. By default, model_selection_cv is False.
model_selection_n_folds (int): The number of folds that should be used for the cross validation grid search if model_selection_cv is True.
YAML specification:
my_knn_method: KNN: # sklearn parameters (same names as in original sklearn class) weights: uniform # always use this setting for weights n_neighbors: [5, 10, 15] # find the optimal number of neighbors # Additional parameter that determines whether to print convergence warnings show_warnings: True # if any of the parameters under KNN is a list and model_selection_cv is True, # a grid search will be done over the given parameters, using the number of folds specified in model_selection_n_folds, # and the optimal model will be selected model_selection_cv: True model_selection_n_folds: 5 # alternative way to define ML method with default values: my_default_knn: KNN
ProbabilisticBinaryClassifier
ProbabilisticBinaryClassifier predicts the class assignment in binary classification case based on encoding examples by number of successful trials and total number of trials. It models this ratio by one beta distribution per class and predicts the class of the new examples using log-posterior odds ratio with threshold at 0.
ProbabilisticBinaryClassifier is based on the paper (details on the classification can be found in the Online Methods section): Emerson, Ryan O., William S. DeWitt, Marissa Vignali, Jenna Gravley, Joyce K. Hu, Edward J. Osborne, Cindy Desmarais, et al. ‘Immunosequencing Identifies Signatures of Cytomegalovirus Exposure History and HLA-Mediated Effects on the T Cell Repertoire’. Nature Genetics 49, no. 5 (May 2017): 659–65. doi.org/10.1038/ng.3822.
Arguments:
max_iterations (int): maximum number of iterations while optimizing the parameters of the beta distribution (same for both classes)
update_rate (float): how much the computed gradient should influence the updated value of the parameters of the beta distribution
likelihood_threshold (float): at which threshold to stop the optimization (default -1e-10)
YAML specification:
my_probabilistic_classifier: # user-defined name of the ML method ProbabilisticBinaryClassifier: # method name max_iterations: 1000 update_rate: 0.01
RandomForestClassifier
This is a wrapper of scikit-learn’s RandomForestClassifier class. Please see the scikit-learn documentation of RandomForestClassifier for the parameters.
Note: if you are interested in plotting the coefficients of the random forest classifier model, consider running the Coefficients report.
Scikit-learn models can be trained in two modes:
1. Creating a model using a given set of hyperparameters, and relying on the selection and assessment loop in the TrainMLModel instruction to select the optimal model.
2. Passing a range of different hyperparameters to RandomForestClassifier, and using a third layer of nested cross-validation to find the optimal hyperparameters through grid search. In this case, only the RandomForestClassifier model with the optimal hyperparameter settings is further used in the inner selection loop of the TrainMLModel instruction.
By default, mode 1 is used. In order to use mode 2, model_selection_cv and model_selection_n_folds must be set.
Arguments:
RandomForestClassifier (dict): Under this key, hyperparameters can be specified that will be passed to the scikit-learn class. Any scikit-learn hyperparameters can be specified here. In mode 1, a single value must be specified for each of the scikit-learn hyperparameters. In mode 2, it is possible to specify a range of different hyperparameters values in a list. It is also allowed to mix lists and single values in mode 2, in which case the grid search will only be done for the lists, while the single-value hyperparameters will be fixed. In addition to the scikit-learn hyperparameters, parameter show_warnings (True/False) can be specified here. This determines whether scikit-learn warnings, such as convergence warnings, should be printed. By default show_warnings is True.
model_selection_cv (bool): If any of the hyperparameters under RandomForestClassifier is a list and model_selection_cv is True, a grid search will be done over the given hyperparameters, using the number of folds specified in model_selection_n_folds. By default, model_selection_cv is False.
model_selection_n_folds (int): The number of folds that should be used for the cross validation grid search if model_selection_cv is True.
YAML specification:
my_random_forest_classifier: # user-defined method name RandomForestClassifier: # name of the ML method # sklearn parameters (same names as in original sklearn class) random_state: 100 # always use this value for random state n_estimators: [10, 50, 100] # find the optimal number of trees in the forest # Additional parameter that determines whether to print convergence warnings show_warnings: True # if any of the parameters under RandomForestClassifier is a list and model_selection_cv is True, # a grid search will be done over the given parameters, using the number of folds specified in model_selection_n_folds, # and the optimal model will be selected model_selection_cv: True model_selection_n_folds: 5 # alternative way to define ML method with default values: my_default_random_forest: RandomForestClassifier
ReceptorCNN
A CNN which separately detects motifs using CNN kernels in each chain of paired receptor data, combines the kernel activations into a unique representation of the receptor and uses this representation to predict the antigen binding.
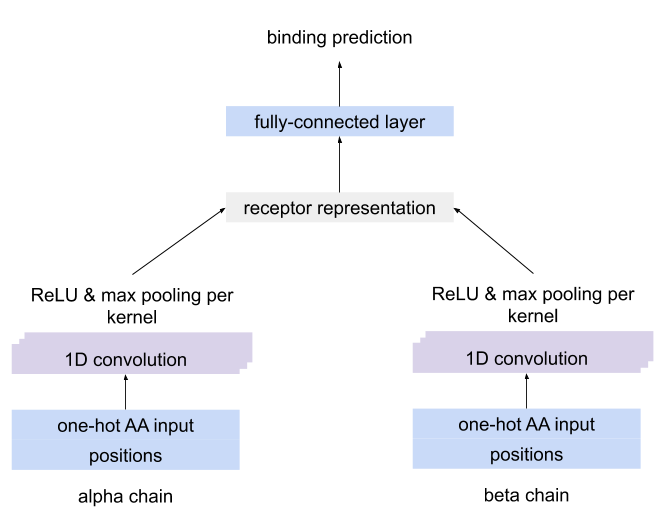
The architecture of the CNN for paired-chain receptor data
Requires one-hot encoded data as input (as produced by OneHot encoder).
Notes:
ReceptorCNN can only be used with ReceptorDatasets, it does not work with SequenceDatasets
ReceptorCNN can only be used for binary classification, not multi-class classification.
Arguments:
kernel_count (count): number of kernels that will look for motifs for one chain
kernel_size (list): sizes of the kernels = how many amino acids to consider at the same time in the chain sequence, can be a tuple of values; e.g. for value [3, 4] of kernel_size, kernel_count*len(kernel_size) kernels will be created, with kernel_count kernels of size 3 and kernel_count kernels of size 4 per chain
positional_channels (int): how many positional channels where included in one-hot encoding of the receptor sequences (default is 3 in one-hot encoder)
sequence_type (SequenceType): type of the sequence
device: which device to use for the model (cpu or gpu) - for more details see PyTorch documentation on device parameter
number_of_threads (int): how many threads to use
random_seed (int): number used as a seed for random initialization
learning_rate (float): learning rate scaling the step size for optimization algorithm
iteration_count (int): for how many iterations to train the model
l1_weight_decay (float): weight decay l1 value for the CNN; encourages sparser representations
l2_weight_decay (float): weight decay l2 value for the CNN; shrinks weight coefficients towards zero
batch_size (int): how many receptors to process at once
training_percentage (float): what percentage of data to use for training (the rest will be used for validation); values between 0 and 1
evaluate_at (int): when to evaluate the model, e.g. every 100 iterations
background_probabilities: used for rescaling the kernel values to produce information gain matrix; represents the background probability of each amino acid (without positional information); if not specified, uniform background is assumed
YAML specification:
my_receptor_cnn: ReceptorCNN: kernel_count: 5 kernel_size: [3] positional_channels: 3 sequence_type: amino_acid device: cpu number_of_threads: 16 random_seed: 100 learning_rate: 0.01 iteration_count: 10000 l1_weight_decay: 0 l2_weight_decay: 0 batch_size: 5000
SVC
This is a wrapper of scikit-learn’s LinearSVC class. Please see the scikit-learn documentation of SVC for the parameters.
Note: if you are interested in plotting the coefficients of the SVC model, consider running the Coefficients report.
Scikit-learn models can be trained in two modes:
1. Creating a model using a given set of hyperparameters, and relying on the selection and assessment loop in the TrainMLModel instruction to select the optimal model.
2. Passing a range of different hyperparameters to SVC, and using a third layer of nested cross-validation to find the optimal hyperparameters through grid search. In this case, only the SVC model with the optimal hyperparameter settings is further used in the inner selection loop of the TrainMLModel instruction.
By default, mode 1 is used. In order to use mode 2, model_selection_cv and model_selection_n_folds must be set.
Arguments:
SVC (dict): Under this key, hyperparameters can be specified that will be passed to the scikit-learn class. Any scikit-learn hyperparameters can be specified here. In mode 1, a single value must be specified for each of the scikit-learn hyperparameters. In mode 2, it is possible to specify a range of different hyperparameters values in a list. It is also allowed to mix lists and single values in mode 2, in which case the grid search will only be done for the lists, while the single-value hyperparameters will be fixed. In addition to the scikit-learn hyperparameters, parameter show_warnings (True/False) can be specified here. This determines whether scikit-learn warnings, such as convergence warnings, should be printed. By default show_warnings is True.
model_selection_cv (bool): If any of the hyperparameters under SVC is a list and model_selection_cv is True, a grid search will be done over the given hyperparameters, using the number of folds specified in model_selection_n_folds. By default, model_selection_cv is False.
model_selection_n_folds (int): The number of folds that should be used for the cross validation grid search if model_selection_cv is True.
YAML specification:
my_svc: # user-defined method name SVC: # name of the ML method # sklearn parameters (same names as in original sklearn class) C: [0.01, 0.1, 1, 10, 100] # find the optimal value for C # Additional parameter that determines whether to print convergence warnings show_warnings: True # if any of the parameters under SVC is a list and model_selection_cv is True, # a grid search will be done over the given parameters, using the number of folds specified in model_selection_n_folds, # and the optimal model will be selected model_selection_cv: True model_selection_n_folds: 5 # alternative way to define ML method with default values: my_default_svc: SVC
SVM
This is a wrapper of scikit-learn’s SVC class. Please see the scikit-learn documentation of SVC for the parameters.
Note: if you are interested in plotting the coefficients of the SVM model, consider running the Coefficients report.
Scikit-learn models can be trained in two modes:
1. Creating a model using a given set of hyperparameters, and relying on the selection and assessment loop in the TrainMLModel instruction to select the optimal model.
2. Passing a range of different hyperparameters to SVM, and using a third layer of nested cross-validation to find the optimal hyperparameters through grid search. In this case, only the SVM model with the optimal hyperparameter settings is further used in the inner selection loop of the TrainMLModel instruction.
By default, mode 1 is used. In order to use mode 2, model_selection_cv and model_selection_n_folds must be set.
Arguments:
SVM (dict): Under this key, hyperparameters can be specified that will be passed to the scikit-learn class. Any scikit-learn hyperparameters can be specified here. In mode 1, a single value must be specified for each of the scikit-learn hyperparameters. In mode 2, it is possible to specify a range of different hyperparameters values in a list. It is also allowed to mix lists and single values in mode 2, in which case the grid search will only be done for the lists, while the single-value hyperparameters will be fixed. In addition to the scikit-learn hyperparameters, parameter show_warnings (True/False) can be specified here. This determines whether scikit-learn warnings, such as convergence warnings, should be printed. By default show_warnings is True.
model_selection_cv (bool): If any of the hyperparameters under SVM is a list and model_selection_cv is True, a grid search will be done over the given hyperparameters, using the number of folds specified in model_selection_n_folds. By default, model_selection_cv is False.
model_selection_n_folds (int): The number of folds that should be used for the cross validation grid search if model_selection_cv is True.
YAML specification:
my_svm: # user-defined method name SVM: # name of the ML method # sklearn parameters (same names as in original sklearn class) C: [0.01, 0.1, 1, 10, 100] # find the optimal value for C kernel: linear # Additional parameter that determines whether to print convergence warnings show_warnings: True # if any of the parameters under SVM is a list and model_selection_cv is True, # a grid search will be done over the given parameters, using the number of folds specified in model_selection_n_folds, # and the optimal model will be selected model_selection_cv: True model_selection_n_folds: 5 # alternative way to define ML method with default values: my_default_svm: SVM
TCRdistClassifier
Implementation of a nearest neighbors classifier based on TCR distances as presented in Dash P, Fiore-Gartland AJ, Hertz T, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017; 547(7661):89-93. doi:10.1038/nature22383.
This method is implemented using scikit-learn’s KNeighborsClassifier with k determined at runtime from the training dataset size and weights linearly scaled to decrease with the distance of examples.
Arguments:
percentage (float): percentage of nearest neighbors to consider when determining receptor specificity based on known receptors (between 0 and 1)
show_warnings (bool): whether to show warnings generated by scikit-learn, by default this is True.
YAML specification:
my_tcr_method: TCRdistClassifier: percentage: 0.1 show_warnings: True
Preprocessings
ChainRepertoireFilter
Removes all repertoires from the RepertoireDataset object which contain at least one sequence with chain different than “keep_chain” parameter. Note that this filter filters out repertoires, not individual sequences, and can thus only be applied to RepertoireDatasets.
Since the filter removes repertoires from the dataset (examples in machine learning setting), it cannot be used with TrainMLModel instruction. If you want to filter out repertoires including a given chain, see DatasetExport instruction with preprocessing.
Arguments:
keep_chain (
SequenceType): Which chain should be kept.YAML specification:
preprocessing_sequences: my_preprocessing: - my_filter: ChainRepertoireFilter: keep_chain: TRB
ClonesPerRepertoireFilter
Removes all repertoires from the RepertoireDataset, which contain fewer clonotypes than specified by the lower_limit, or more clonotypes than specified by the upper_limit. Note that this filter filters out repertoires, not individual sequences, and can thus only be applied to RepertoireDatasets.
Since the filter removes repertoires from the dataset (examples in machine learning setting), it cannot be used with TrainMLModel instruction. If you want to use this filter, see DatasetExport instruction with preprocessing.
Arguments:
lower_limit (int): The minimal inclusive lower limit for the number of clonotypes allowed in a repertoire.
upper_limit (int): The maximal inclusive upper limit for the number of clonotypes allowed in a repertoire.
When no lower or upper limit is specified, or the value -1 is specified, the limit is ignored.
YAML specification:
preprocessing_sequences: my_preprocessing: - my_filter: ClonesPerRepertoireFilter: lower_limit: 100 upper_limit: 100000
CountPerSequenceFilter
Removes all sequences from a Repertoire when they have a count below low_count_limit, or sequences with no count value if remove_without_counts is True. This filter can be applied to Repertoires and RepertoireDatasets.
Arguments:
low_count_limit (int): The inclusive minimal count value in order to retain a given sequence.
remove_without_count (bool): Whether the sequences without a reported count value should be removed.
remove_empty_repertoires (bool): Whether repertoires without sequences should be removed. Only has an effect when remove_without_count is also set to True. If this is true, this preprocessing cannot be used with TrainMLModel instruction, but only with DatasetExport instruction instead.
batch_size (int): number of repertoires that can be loaded at the same time (only affects the speed when applying this filter on a RepertoireDataset)
YAML specification:
preprocessing_sequences: my_preprocessing: - my_filter: CountPerSequenceFilter: remove_without_count: True remove_empty_repertoires: True low_count_limit: 3 batch_size: 4
DuplicateSequenceFilter
Collapses duplicate nucleotide or amino acid sequences within each repertoire in the given RepertoireDataset. This filter can be applied to Repertoires and RepertoireDatasets.
Sequences are considered duplicates if the following fields are identical:
amino acid or nucleotide sequence (whichever is specified)
v and j genes (note that the full field including subgroup + gene is used for matching, i.e. V1 and V1-1 are not considered duplicates)
chain
region type
For all other fields (the non-specified sequence type, custom lists, sequence identifier) only the first occurring value is kept.
Note that this means the count value of a sequence with a given sequence identifier might not be the same as before removing duplicates, unless count_agg = FIRST is used.
Arguments:
filter_sequence_type (
SequenceType): Whether the sequences should be collapsed on the nucleotide or amino acid level. Valid values are: [‘AMINO_ACID’, ‘NUCLEOTIDE’].batch_size (int): number of repertoires that can be loaded at the same time (only affects the speed)
count_agg (
CountAggregationFunction): determines how the sequence counts of duplicate sequences are aggregated. Valid values are: [‘SUM’, ‘MAX’, ‘MIN’, ‘MEAN’, ‘FIRST’, ‘LAST’].YAML specification:
preprocessing_sequences: my_preprocessing: - my_filter: DuplicateSequenceFilter: # required parameters: filter_sequence_type: AMINO_ACID # optional parameters (if not specified the values bellow will be used): batch_size: 4 count_agg: SUM
MetadataRepertoireFilter
Removes repertoires from a RepertoireDataset based on information stored in the metadata_file. Note that this filters out repertoires, not individual sequences, and can thus only be applied to RepertoireDatasets.
Since this filter changes the number of repertoires (examples for the machine learning task), it cannot be used with TrainMLModel instruction. To filter out repertoires, use preprocessing from the DatasetExport instruction that will create a new dataset ready to be used for training machine learning models.
Arguments:
criteria (dict): a nested dictionary that specifies the criteria for keeping certain columns. See
CriteriaMatcherfor a more detailed explanation.YAML specification:
preprocessing_sequences: my_preprocessing: - my_filter: # Example filter that keeps repertoires with values greater than 1 in the "my_column_name" column of the metadata_file MetadataRepertoireFilter: type: GREATER_THAN value: type: COLUMN name: my_column_name threshold: 1
SubjectRepertoireCollector
Merges all the Repertoires in a RepertoireDataset that have the same ‘subject_id’ specified in the metadata. The result is a RepertoireDataset with one Repertoire per subject. This preprocessing cannot be used in combination with TrainMLModel instruction because it can change the number of examples. To combine the repertoires in this way, use this preprocessing with DatasetExport instruction.
YAML specification:
preprocessing_sequences: my_preprocessing: - my_filter: SubjectRepertoireCollector
Instructions
DatasetExport
DatasetExport instruction takes a list of datasets as input, optionally applies preprocessing steps, and outputs the data in specified formats.
Arguments:
datasets (list): a list of datasets to export in all given formats
preprocessing_sequence (str): which preprocessing sequence to use on the dataset(s), this item is optional and does not have to be specified. When specified, the same preprocessing sequence will be applied to all datasets.
formats (list): a list of formats in which to export the datasets. Valid values are: ImmuneML, AIRR.
YAML specification:
my_dataset_export_instruction: # user-defined instruction name type: DatasetExport # which instruction to execute datasets: # list of datasets to export - my_generated_dataset - my_dataset_from_adaptive preprocessing_sequence: my_preprocessing_sequence export_formats: # list of formats to export the datasets to - AIRR - ImmuneML
ExploratoryAnalysis
Allows exploratory analysis of different datasets using encodings and reports.
Analysis is defined by a dictionary of ExploratoryAnalysisUnit objects that encapsulate a dataset, an encoding [optional] and a report to be executed on the [encoded] dataset. Each analysis specified under analyses is completely independent from all others.
Arguments:
analyses (dict): a dictionary of analyses to perform. The keys are the names of different analyses, and the values for each of the analyses are:
dataset: dataset on which to perform the exploratory analysis
preprocessing_sequence: which preprocessings to use on the dataset, this item is optional and does not have to be specified.
encoding: how to encode the dataset before running the report, this item is optional and does not have to be specified.
labels: if encoding is specified, the relevant labels must be specified here.
report: which report to run on the dataset. Reports specified here may be of the category Data reports or Encoding reports, depending on whether ‘encoding’ was specified.
number_of_processes: (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
YAML specification:
my_expl_analysis_instruction: # user-defined instruction name type: ExploratoryAnalysis # which instruction to execute analyses: # analyses to perform my_first_analysis: # user-defined name of the analysis dataset: d1 # dataset to use in the first analysis preprocessing_sequence: p1 # preprocessing sequence to use in the first analysis report: r1 # which report to generate using the dataset d1 my_second_analysis: # user-defined name of another analysis dataset: d1 # dataset to use in the second analysis - can be the same or different from other analyses encoding: e1 # encoding to apply on the specified dataset (d1) report: r2 # which report to generate in the second analysis labels: # labels present in the dataset d1 which will be included in the encoded data on which report r2 will be run - celiac # name of the first label as present in the column of dataset's metadata file - CMV # name of the second label as present in the column of dataset's metadata file number_of_processes: 4 # number of parallel processes to create (could speed up the computation)
MLApplication
Instruction which enables using trained ML models and encoders on new datasets which do not necessarily have labeled data.
The predictions are stored in the predictions.csv in the result path in the following format:
example_id
cmv
cmv_true_proba
cmv_false_proba
e1
True
0.8
0.2
e2
False
0.2
0.8
e3
True
0.78
0.22
Arguments:
dataset: dataset for which examples need to be classified
config_path: path to the zip file exported from MLModelTraining instruction (which includes train ML model, encoder, preprocessing etc.)
number_of_processes (int): number of processes to use for prediction
Specification example for the MLApplication instruction:
instruction_name: type: MLApplication dataset: d1 config_path: ./config.zip number_of_processes: 4
Simulation
A simulation is an instruction that implants synthetic signals into the given dataset according to given parameters. This results in a new dataset containing modified sequences, and is annotated with metadata labels according to the implanted signals.
Arguments:
dataset: original dataset which will be used as a basis for implanting signals from the simulation
simulation: definition of how to perform the simulation.
export_formats: in which formats to export the dataset after simulation. Valid values are: ImmuneML, AIRR. Important note: Binary files in ImmuneML might not be compatible between different immuneML versions.
YAML specification:
my_simulation_instruction: # user-defined name of the instruction type: Simulation # which instruction to execute dataset: my_dataset # which dataset to use for implanting the signals simulation: my_simulation # how to implanting the signals - definition of the simulation export_formats: [AIRR] # in which formats to export the dataset
Subsampling
Subsampling is an instruction that subsamples a given dataset and creates multiple smaller dataset according to the parameters provided.
Arguments:
dataset (Dataset): original dataset which will be used as a basis for subsampling
subsampled_dataset_sizes (list): a list of dataset sizes (number of examples) each subsampled dataset should have
dataset_export_formats (list): in which formats to export the subsampled datasets. Valid values are: ImmuneML, AIRR.
YAML specification:
my_subsampling_instruction: # user-defined name of the instruction type: Subsampling # which instruction to execute dataset: my_dataset # original dataset to be subsampled, with e.g., 300 examples subsampled_dataset_sizes: # how large the subsampled datasets should be, one dataset will be created for each list item - 200 # one subsampled dataset with 200 examples (200 repertoires if my_dataset was repertoire dataset) - 100 # the other subsampled dataset will have 100 examples dataset_export_formats: # in which formats to export the subsampled datasets - ImmuneML - AIRR
TrainMLModel
Class implementing hyperparameter optimization and training and assessing the model through nested cross-validation (CV). The process is defined by two loops:
the outer loop over defined splits of the dataset for performance assessment
the inner loop over defined hyperparameter space and with cross-validation or train & validation split to choose the best hyperparameters.
Optimal model chosen by the inner loop is then retrained on the whole training dataset in the outer loop.
Note: If you are interested in plotting the performance of all combinations of encodings and ML methods on the test set, consider running the MLSettingsPerformance report as hyperparameter report in the assessment loop.
Arguments:
dataset: dataset to use for training and assessing the classifier
strategy: how to search different hyperparameters; common options include grid search, random search. Valid values are: GridSearch.
settings (list): a list of combinations of preprocessing_sequence, encoding and ml_method. preprocessing_sequence is optional, while encoding and ml_method are mandatory. These three options (and their parameters) can be optimized over, choosing the highest performing combination.
assessment: description of the outer loop (for assessment) of nested cross-validation. It describes how to split the data, how many splits to make, what percentage to use for training and what reports to execute on those splits. See SplitConfig.
selection: description of the inner loop (for selection) of nested cross-validation. The same as assessment argument, just to be executed in the inner loop. See SplitConfig.
metrics (list): a list of metrics (accuracy, balanced_accuracy, confusion_matrix, f1_micro, f1_macro, f1_weighted, precision, recall, auc, log_loss) to compute for all splits and settings created during the nested cross-validation. These metrics will be computed only for reporting purposes. For choosing the optimal setting, optimization_metric will be used.
optimization_metric: a metric to use for optimization (one of accuracy, balanced_accuracy, confusion_matrix, f1_micro, f1_macro, f1_weighted, precision, recall, auc, log_loss) and assessment in the nested cross-validation.
labels (list): a list of labels for which to train the classifiers. The goal of the nested CV is to find the setting which will have best performance in predicting the given label (e.g., if a subject has experienced an immune event or not). Performance and optimal settings will be reported for each label separately. If a label is binary, instead of specifying only its name, one should explicitly set the name of the positive class as well under parameter positive_class. If positive class is not set, one of the label classes will be assumed to be positive.
number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
reports (list): a list of report names to be executed after the nested CV has finished to show the overall performance or some statistic; the reports that can be provided here are Train ML model reports.
refit_optimal_model (bool): if the final combination of preprocessing-encoding-ML model should be refitted on the full dataset thus providing the final model to be exported from instruction; alternatively, train combination from one of the assessment folds will be used
YAML specification:
my_nested_cv_instruction: # user-defined name of the instruction type: TrainMLModel # which instruction should be executed settings: # a list of combinations of preprocessing, encoding and ml_method to optimize over - preprocessing: seq1 # preprocessing is optional encoding: e1 # mandatory field ml_method: simpleLR # mandatory field - preprocessing: seq1 # the second combination encoding: e2 ml_method: simpleLR assessment: # outer loop of nested CV split_strategy: random # perform Monte Carlo CV (randomly split the data into train and test) split_count: 1 # how many train/test datasets to generate training_percentage: 0.7 # what percentage of the original data should be used for the training set reports: # reports to execute on training/test datasets, encoded datasets and trained ML methods data_splits: # list of reports to execute on training/test datasets (before they are encoded) - rep1 encoding: # list of reports to execute on encoded training/test datasets - rep2 models: # list of reports to execute on trained ML methods for each assessment CV split - rep3 selection: # inner loop of nested CV split_strategy: k_fold # perform k-fold CV split_count: 5 # how many fold to create: here these two parameters mean: do 5-fold CV reports: data_splits: # list of reports to execute on training/test datasets (in the inner loop, so these are actually training and validation datasets) - rep1 models: # list of reports to execute on trained ML methods for each selection CV split - rep2 encoding: # list of reports to execute on encoded training/test datasets (again, it is training/validation here) - rep3 labels: # list of labels to optimize the classifier for, as given in the metadata for the dataset - celiac: positive_class: + # if it's binary classification, positive class parameter should be set - T1D # this is not binary label, so no need to specify positive class dataset: d1 # which dataset to use for the nested CV strategy: GridSearch # how to choose the combinations which to test from settings (GridSearch means test all) metrics: # list of metrics to compute for all settings, but these do not influence the choice of optimal model - accuracy - auc reports: # list of reports to execute when nested CV is finished to show overall performance - rep4 number_of_processes: 4 # number of parallel processes to create (could speed up the computation) optimization_metric: balanced_accuracy # the metric to use for choosing the optimal model and during training refit_optimal_model: False # use trained model, do not refit on the full dataset
SplitConfig
SplitConfig describes how to split the data for cross-validation. It allows for the following combinations:
loocv (leave-one-out cross-validation)
k_fold (k-fold cross-validation)
stratified_k_fold (stratified k-fold cross-validation that can be used when immuneML is used for single-label classification, see `this documentation<https://scikit-learn.org/stable/modules/generated/sklearn.model_selection.StratifiedKFold.html>`_ for more details on how this is implemented)
random (Monte Carlo cross-validation - randomly splitting the dataset to training and test datasets)
manual (train and test dataset are explicitly specified by providing metadata files for the two datasets - currently available only for repertoire datasets)
leave_one_out_stratification (leave-one-out CV where one refers to a specific parameter, e.g. if subject is known in a receptor dataset, it is possible to have leave-subject-out CV - currently only available for receptor datasets).
Arguments:
split_strategy: one of the types of cross-validation listed above (LOOCV, K_FOLD, STRATIFIED_K_FOLD, MANUAL, `` or RANDOM)
split_count (int): if split_strategy is K_FOLD, then this defined how many splits to make (K), if split_strategy is RANDOM, split_count defines how many random splits to make, resulting in split_count training/test dataset pairs, or if split_strategy is LOOCV, MANUAL or LEAVE_ONE_OUT_STRATIFICATION, split_count does not need to be specified.
training_percentage: if split_strategy is RANDOM, this defines which portion of the original dataset to use for creating the training dataset; for other values of split_strategy, this parameter is not used.
reports: defines which reports to execute on which datasets or settings. See ReportConfig for more details.
manual_config: if split strategy is MANUAL, here the paths to metadata files should be given (fields train_metadata_path and test_metadata_path). The matching of examples is done using the “subject_id” field so it has to be present in both the original dataset and the metadata files provided here. Manual splitting to train and test dataset is currently supported only for repertoire datasets. If split strategy is anything else, this field has no effect and can be omitted.
leave_one_out_config: if split strategy is LEAVE_ONE_OUT_STRATIFICATION, this config describes which parameter to use for stratification thus making a list of train/test dataset combinations in which in the test set there are examples with only one value of the specified parameter. leave_one_out_config argument accepts two inputs: parameter which is the name of the parameter to use for stratification and min_count which defines the minimum number of examples that can be present in the test dataset. This type of generating train and test datasets is only supported for receptor datasets so far. If split strategy is anything else, this field has no effect and can be omitted.
YAML specification:
# as a part of a TrainMLModel instruction, defining the outer (assessment) loop of nested cross-validation: assessment: # outer loop of nested CV split_strategy: random # perform Monte Carlo CV (randomly split the data into train and test) split_count: 5 # how many train/test datasets to generate training_percentage: 0.7 # what percentage of the original data should be used for the training set reports: # reports to execute on training/test datasets, encoded datasets and trained ML methods data_splits: # list of data reports to execute on training/test datasets (before they are encoded) - rep1 encoding: # list of encoding reports to execute on encoded training/test datasets - rep2 models: # list of ML model reports to execute on the trained classifiers in the assessment loop - rep3 # as a part of a TrainMLModel instruction, defining the inner (selection) loop of nested cross-validation: selection: # inner loop of nested CV split_strategy: leave_one_out_stratification leave_one_out_config: # perform leave-(subject)-out CV parameter: subject # which parameter to use for splitting, must be present in the metadata for each example min_count: 1 # what is the minimum number of examples with unique value of the parameter specified above for the analysis to be valid reports: # reports to execute on training/test datasets, encoded datasets and trained ML methods data_splits: # list of data reports to execute on training/test datasets (before they are encoded) - rep1 encoding: # list of encoding reports to execute on encoded training/test datasets - rep2 encoding: # list of ML model reports to execute the trained classifiers in the selection loop - rep3
ReportConfig
A class encapsulating different report lists which can be executed while performing nested cross-validation (CV) using TrainMLModel instruction. All arguments are optional.
Arguments:
data: Data reports to be executed on the whole dataset before it is split to training/test or training/validation
data_splits: Data reports to be executed after the data has been split into training and test (assessment CV loop) or training and validation (selection CV loop) datasets before they are encoded
models: ML model reports to be executed on all trained classifiers
encoding: Encoding reports to be executed on each of the encoded training/test datasets or training/validation datasets
YAML specification:
# as a part of a TrainMLModel instruction, defining the outer (assessment) loop of nested cross-validation: assessment: # outer loop of nested CV split_strategy: random # perform Monte Carlo CV (randomly split the data into train and test) split_count: 5 # how many train/test datasets to generate training_percentage: 0.7 # what percentage of the original data should be used for the training set reports: # reports to execute on training/test datasets, encoded datasets and trained ML methods data_splits: # list of reports to execute on training/test datasets (before they are preprocessed and encoded) - my_data_split_report encoding: # list of reports to execute on encoded training/test datasets - my_encoding_report # as a part of a TrainMLModel instruction, defining the inner (selection) loop of nested cross-validation: selection: # inner loop of nested CV split_strategy: random # perform Monte Carlo CV (randomly split the data into train and validation) split_count: 5 # how many train/validation datasets to generate training_percentage: 0.7 # what percentage of the original data should be used for the training set reports: # reports to execute on training/validation datasets, encoded datasets and trained ML methods data_splits: # list of reports to execute on training/validation datasets (before they are preprocessed and encoded) - my_data_split_report encoding: # list of reports to execute on encoded training/validation datasets - my_encoding_report models: - my_ml_model_report