How to specify an analysis with YAML¶
Analyses in immuneML are specified through a YAML specification file with a fixed structure. Depending on the specification, immuneML can execute different tasks, such as training ML models for receptor or repertoire classification, simulate data or perform exploratory analyses. For all the options that can be specified, see YAML specification.
Structure of the analysis specification¶
The analysis specification consists of three main parts: definitions, instructions and output.
Specifying Definitions¶
definitions refer to components, which will be used within the instructions. They include:
datasets: specifying where data is located, what format the data is in, and how it should be imported (see How to import data into immuneML for more details),preprocessing_sequences: defining one or more preprocessing steps to be taken on the dataset (this is optional),encodings: different data representations,ml_methods: different machine learning methods (e.g., SVM or KNN),reports: specific plots or statistics to apply to the raw or encoded data, ML methods or results.
Simulation-specific components (only relevant when running a Simulation instruction) are:
motifs: parts of the simulation definition defined by a seed and a way to create specific motif instances from the seed,signals: parts of the simulation which can include multiple motifs and correspond to a single label for subsequent classification tasks,simulations: define how to combine different signals and how to implant them in the dataset.
Under definitions, each analysis sub-component is defined using a user-specifiable key. In the examples below, we will use the prefix ‘my_’ to identify these keys, but in practice it is possible to specify any kind of name here. These keys are unique names that identify the settings for a component, and they are later on referenced in the instructions (for example: to specify which of the imported datasets to use in a given instruction).
The import of two datasets may be defined as follows:
definitions:
datasets:
my_repertoire_dataset: # user-defined key for the first dataset
format: AIRR # import of a repertoire dataset
params:
path: path/to/first/data/
metadata_file: path/to/first/metadata.csv
my_receptor_dataset: # user-defined key for the second dataset
format: AIRR # import of a receptor dataset
params:
path: path/to/second/data/
is_repertoire: false
paired: true
receptor_chains: TRA_TRB
metadata_column_mapping: # map column names of the file to label names
epitope_column_name: my_epitope # my_epitope can be used as label
Where the imported datasets can under instructions be referenced using the keys my_repertoire_dataset and my_receptor_dataset.
Note that in practice, most analyses use just one dataset.
An example of a full definitions section which may be used for a machine learning task is given below.
See also How to train and assess a receptor or repertoire-level ML classifier for more details.
definitions:
datasets: # every instruction uses a dataset
my_dataset:
format: AIRR
params:
path: path/to/data/
metadata_file: path/to/metadata.csv
preprocessing_sequences:
my_preprocessing:
- my_beta_chain_filter:
ChainRepertoireFilter:
keep_chain: TRB
ml_methods:
my_log_reg: LogisticRegression
my_svm: SVM
encodings:
my_kmer_freq_encoding_1: KmerFrequency # KmerFrequency with default parameters
my_kmer_freq_encoding_2: # KmerFrequency with user-defined parameters
KmerFrequency:
k: 5
reports:
my_seq_length_distribution: SequenceLengthDistribution
The definitions section used for Simulation contains different components, as shown in the example below.
See also How to simulate antigen or disease-associated signals in AIRR datasets for more details.
definitions:
datasets: # every instruction uses a dataset
my_dataset:
format: AIRR
params:
path: path/to/data/
metadata_file: path/to/metadata.csv
motifs:
my_simple_motif:
seed: AAA
instantiation: GappedKmer
signals:
my_simple_signal:
motifs:
- my_simple_motif
implanting: HealthySequence
simulations:
my_simulation:
my_implanting:
signals:
- my_simple_signal
dataset_implanting_rate: 0.5
repertoire_implanting_rate: 0.1
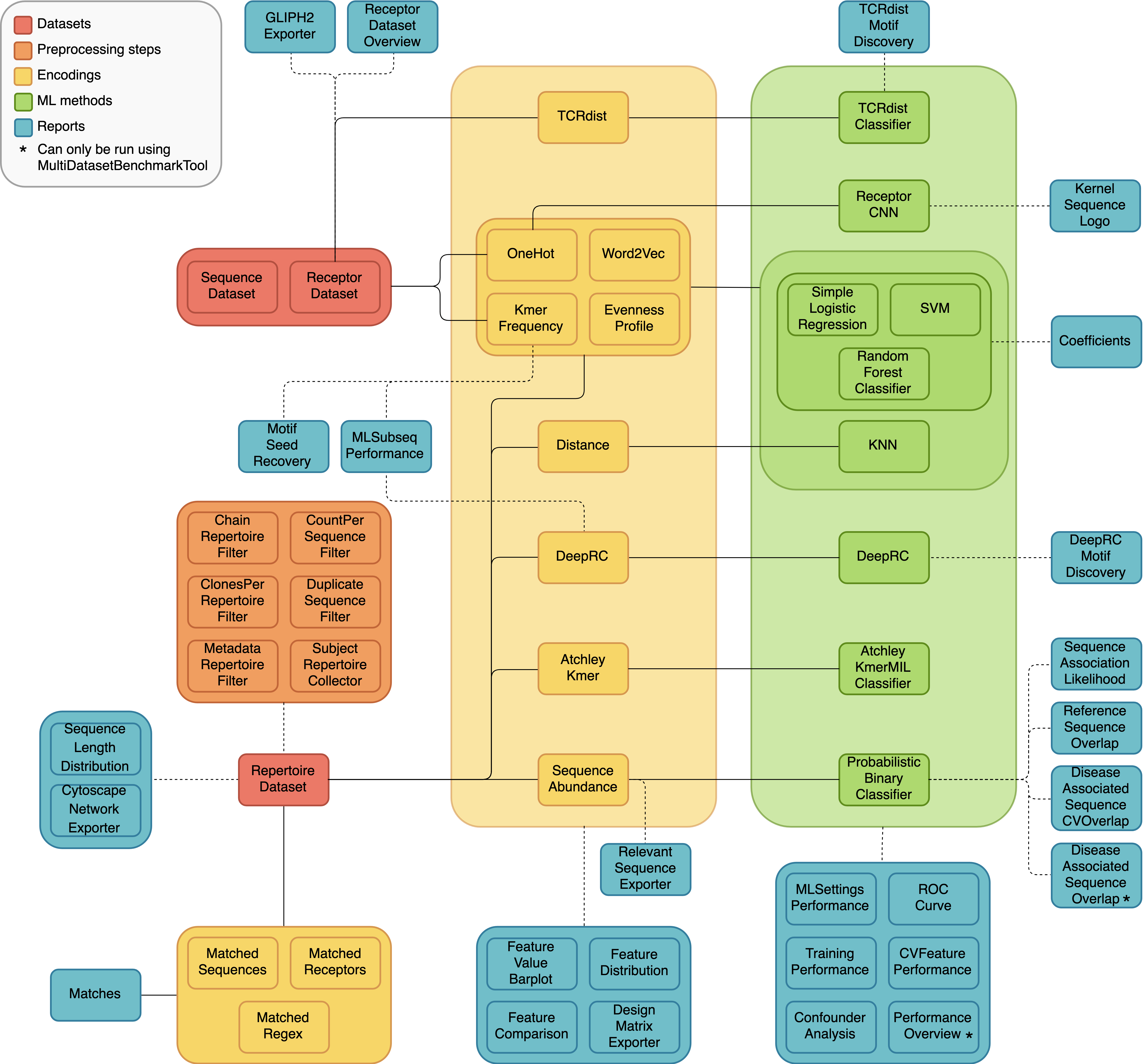
A diagram of all the different dataset types, preprocessing steps, encodings, ML methods and reports, and how they can be combined in different analyses is shown below. The solid lines represent components that are intended to be used together, and the dashed lines indicate optional combinations.
Specifying Instructions¶
Similarly to analysis components, instructions are defined under a user-specifiable key.
Under this key, you should define the instruction type, which defines the type
of analysis that will be done. All other settings are instruction-specific.
Some of the possible instruction types are (see Instructions for the complete list):
The components defined under definitions can be referenced inside the instruction, but anything generated from the instructions is not available to other instructions. If the output of one instruction needs to be used in another other instruction, two separate immuneML runs need to be made (e.g, running immuneML once with the Simulation instruction to generate a dataset, and subsequently using that dataset as an input to a second immuneML run to train a ML model).
An example of the YAML specification for the TrainMLModel instruction is shown below. See the tutorial How to train and assess a receptor or repertoire-level ML classifier for more explanation behind all settings.
instructions:
my_instruction: # user-defined instruction key
type: TrainMLModel
dataset: my_dataset # reference dataset from definitions
labels: [disease]
settings: # settings are made up of preprocessing (optional), ml_method and encoding
- encoding: my_kmer_freq_encoding_1
ml_method: my_log_reg
- preprocessing: my_preprocessing
encoding: my_kmer_freq_encoding_2
ml_method: my_svm
assessment:
split_strategy: random
split_count: 1
training_percentage: 70
reports:
data_splits: [my_seq_length_distribution]
selection:
split_strategy: k_fold
split_count: 5
strategy: GridSearch
metrics: [accuracy]
optimization_metric: accuracy
reports: null # no reports
refit_optimal_model: False
number_of_processes: 4
Specifying output¶
The third and final section of the YAML specification is output, which currently only supports one format: HTML.
The output section may be omitted from the YAML, but if included, it should look like this:
output:
format: HTML
Putting all parts together¶
An example of a complete YAML specification for training an ML model through nested cross-validation is given here:
definitions:
datasets:
d1:
format: AIRR
params:
metadata_file: path/to/metadata.csv
path: path/to/data/
preprocessing_sequences:
my_preprocessing:
- my_beta_chain_filter:
ChainRepertoireFilter:
keep_chain: TRB
ml_methods:
my_log_reg: LogisticRegression
my_svm: SVM
encodings:
my_kmer_freq_encoding_1: KmerFrequency # KmerFrequency with default parameters
my_kmer_freq_encoding_2: # KmerFrequency with user-defined parameters
KmerFrequency:
k: 5
reports:
my_seq_length_distribution: SequenceLengthDistribution
instructions:
my_instruction: # user-defined instruction key
type: TrainMLModel
dataset: my_dataset # reference dataset from definitions
labels: [disease]
settings: # settings are made up of preprocessing (optional), ml_method and encoding
- encoding: my_kmer_freq_encoding_1
ml_method: my_log_reg
- preprocessing: my_preprocessing
encoding: my_kmer_freq_encoding_2
ml_method: my_svm
assessment:
split_strategy: random
split_count: 1
training_percentage: 70
reports:
data_splits: [my_seq_length_distribution]
selection:
split_strategy: k_fold
split_count: 5
strategy: GridSearch
metrics: [accuracy]
optimization_metric: accuracy
reports: null # no reports
refit_optimal_model: False
number_of_processes: 4
output:
format: HTML
Running the specified analysis¶
To run an instruction via command line with the given YAML specification file:
immune-ml path/to/specification.yaml result/folder/path/
Alternatively, create an ImmuneMLApp object in a Python script and pass it the path parameter to the constructor before calling its run() method as follows:
from source.app.ImmuneMLApp import ImmuneMLApp
app = ImmuneMLApp(specification_path="path/to/specification.yaml", result_path="result/folder/path/")
app.run()