How to make an immuneML dataset in Galaxy¶
In Galaxy, an immuneML dataset is simply a Galaxy collection containing all relevant files (including an optional metadata file). The Create dataset Galaxy tool allows users to import data from various formats and create immuneML datasets in Galaxy. These datasets are in an optimized binary (Pickle) format, which reduces the time needed to read the data into immuneML, and ensures that you can quickly import the dataset into Galaxy tools without having to repeatedly specify the import parameters.
Before creating a dataset, the relevant data files must first be uploaded to the Galaxy interface. This can be done either by uploading files from your local computer (use the ‘Upload file’ tool under the ‘Get local data’ menu), or by fetching remote data from the iReceptor Plus Gateway or VDJdb (see How to import remote AIRR datasets in Galaxy).
The imported immuneML dataset will appear as a history item on the right side of the screen, and can later be selected as input to other tools.
The tool has a simplified and an advanced interface. The simplified interface is fully button-based, and relies on default settings for importing datasets. The advanced interface gives full control over import settings through a YAML specification. In most cases, the simplified interface will suffice.
If your history contains more than 100 files, you may want to consider using a Galaxy collection as input using a Galaxy collection as input.
See also the following example Galaxy histories, showing how to run the tool:
immuneML datasets¶
There exist three types of datasets in immuneML:
Repertoire datasets should be used when making predictions per repertoire, such as predicting a disease state.
Sequence datasets should be used when predicting values for single immune receptor chains, such as antigen specificity.
Receptor datasets are the paired variant of sequence datasets, and should be used to make a prediction for each receptor chain pair.
In order to use a dataset for training ML classifiers, the metadata, which contains prediction labels, needs to be available.
For repertoire datasets, the metadata is supplied through a metadata file. The metadata file is a .csv file which contains
one repertoire (filename) per row, and the metadata labels for that repertoire. (see: What should the metadata file look like?).
Note that only the repertoire files that are present in the metadata file will be imported.
For sequence and receptor datasets the metadata should be available in the columns of the sequence data files. For example, VDJdb files contain columns named ‘Epitope’, ‘Epitope gene’ and ‘Epitope species’. These columns can be specified to serve as metadata columns.
Using the simplified ‘Create dataset’ interface¶
In the simplified interface the user has to select an input file format, dataset type and a list of data files to use. For repertoire datasets, a metadata file must be selected from the history, whereas for sequence- and receptor datasets the names of the columns containing metadata must be specified. The names of the metadata columns are in later analyses available as labels for the sequence and receptor datasets.
In subsequent YAML-based analyses, the dataset created through the simplified interface should be specified like this:
definitions:
datasets:
my_analysis_dataset: # user-defined dataset name
format: Pickle
params:
path: dataset.iml_dataset
Note: if an immuneML dataset history element suddenly gives you errors when you use it as an input to other tools (while it used to work before), it could be due to an immuneML version update. To solve this problem, try to rerun the ‘Create dataset’ tool with the same input files (for example by clicking the ‘Run this job again’ button), and use the new immuneML dataset history element.
Using the advanced ‘Create dataset’ interface¶
When using the advanced interface of the ‘Create dataset’ tool, a YAML specification and a list of files must be filled in. The list of selected files should contain all data files to be imported, and additionally in the case of a repertoire dataset a metadata file.
The YAML specification describes how the dataset should be created from the supplied files. See YAML specification for more details on writing a YAML specification file. For this tool, one Dataset must be specified under definitions, and the DatasetExport instruction must be used.
The DatasetExport instruction can here only be used with one dataset (as defined under definitions) and one export format.
Furthermore, the path parameter does not need to be set. Other than this, the specification is written the same as when running immuneML locally.
A complete YAML specification for a repertoire dataset could look like this:
definitions:
datasets:
my_repertoire_dataset: # user-defined dataset name
format: VDJdb
params:
is_repertoire: True # import a repertoire dataset
metadata_file: metadata.csv # the metadata file is identified by name
# other import parameters may be specified here
instructions:
my_dataset_export_instruction: # user-defined instruction name
type: DatasetExport
datasets: # specify the dataset defined above
- my_repertoire_dataset
export_formats:
# only one format can be specified here and the dataset in this format will be
# available as a Galaxy collection afterwards
- Pickle # Can be AIRR (human-readable) or Pickle (recommended for further Galaxy-analysis)
Alternatively, for a receptor dataset the complete YAML specification may look like this:
definitions:
datasets:
my_receptor_dataset: # user-defined dataset name
format: VDJdb
params:
is_repertoire: False
paired: True # if True, import receptor dataset. If False, import sequence dataset
receptor_chains: TRA_TRB # choose from TRA_TRB, TRG_TRD, IGH_IGL and IGH_IGK
metadata_column_mapping: # VDJdb name: immuneML name
# import VDJdb columns Epitope, Epitope gene and Epitope species, and save them
# in metadata fields epitope, epitope_gene and epitope_species which can be used as labels
Epitope: epitope
Epitope gene: epitope_gene
Epitope species: epitope_species
# other import parameters may be specified here
instructions:
my_dataset_export_instruction: # user-defined instruction name
type: DatasetExport
datasets: # specify the dataset defined above
- my_receptor_dataset
export_formats:
# only one format can be specified here and the dataset in this format will be
# available as a Galaxy collection afterwards
- Pickle # Can be AIRR (human-readable) or Pickle (recommended for further Galaxy-analysis)
Note that the export format specified here will determine how dataset import should be defined in the subsequent YAML specifications for other immuneML Galaxy tools (‘Run immuneML with YAML specification’ and ‘Simulate events in an immune dataset’). The recommended format is Pickle, as it is easiest to specify dataset import from Pickle format. If Pickle is chosen as the export format, the dataset definition for subsequent analyses will look like this:
definitions:
datasets:
my_analysis_dataset: # user-defined dataset name
format: Pickle
params:
# note that my_dataset is the name given earlier in the 'Create dataset' YAML
path: my_dataset.iml_dataset
Alternatively, AIRR format may be specified as it is human-readable. When AIRR format is used, all relevant import parameters need to be specified in subsequent analyses:
definitions:
datasets:
my_analysis_dataset: # user-defined dataset name
format: AIRR
params:
# the same value for is_repertoire and metadata_file must be used as in the first YAML
is_repertoire: True
metadata_file: metadata.csv
# other import parameters may be specified here
Note: if you used the ‘Pickle’ export format and your immuneML dataset history element suddenly gives you errors when you use it as an input to other tools (while it used to work before), it could be due to an immuneML version update. To solve this problem, try to rerun the ‘Create dataset’ tool with the same input files (for example by clicking the ‘Run this job again’ button), and use the new immuneML dataset history element.
Using a Galaxy collection as input¶
When your dataset contains many files, it can be cumbersome to have to click on all the files that should be imported. Alternatively, it is possible to create a Galaxy collection of files and import this collection.
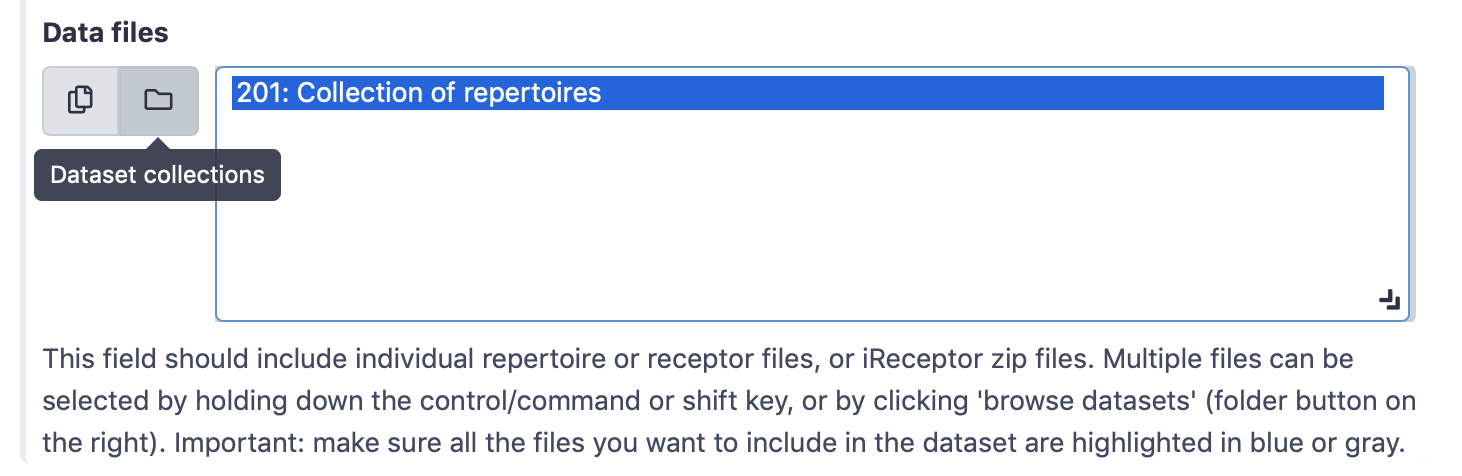
By default, the field ‘Data files’ (simplified interface) or ‘Data and metadata files’ (advanced interface) will show all txt-like files (such as repertoire or receptor files) that are present in the history. By clicking the ‘Dataset collections’ button (folder icon), the menu will instead only show the collections in the history. One or more collections can now be selected.
Tool output¶
This Galaxy tool will produce the following history elements:
Summary: dataset generation: a HTML page describing general characteristics of the dataset, including the name of the dataset (this name should be specified when importing the dataset later in immuneML), the dataset type and size, and a link to download the raw data files.
create_dataset.yaml: the YAML specification file that was used by immuneML to create the dataset. This file can be downloaded and altered (for example to export files in AIRR format, or use non-standard import parameters), and run again using the ‘Advanced’ interface.
immuneML dataset: a Galaxy collection containing the immuneML dataset in Pickle format.