immuneML.reports.data_reports package¶
Submodules¶
immuneML.reports.data_reports.AminoAcidFrequencyDistribution module¶
- class immuneML.reports.data_reports.AminoAcidFrequencyDistribution.AminoAcidFrequencyDistribution(dataset: Dataset = None, alignment: bool = None, relative_frequency: bool = None, split_by_label: bool = None, label: str = None, region_type: RegionType = RegionType.IMGT_CDR3, result_path: Path = None, number_of_processes: int = 1, name: str = None)[source]¶
Bases:
DataReportGenerates a barplot showing the relative frequency of each amino acid at each position in the sequences of a dataset.
Example output:
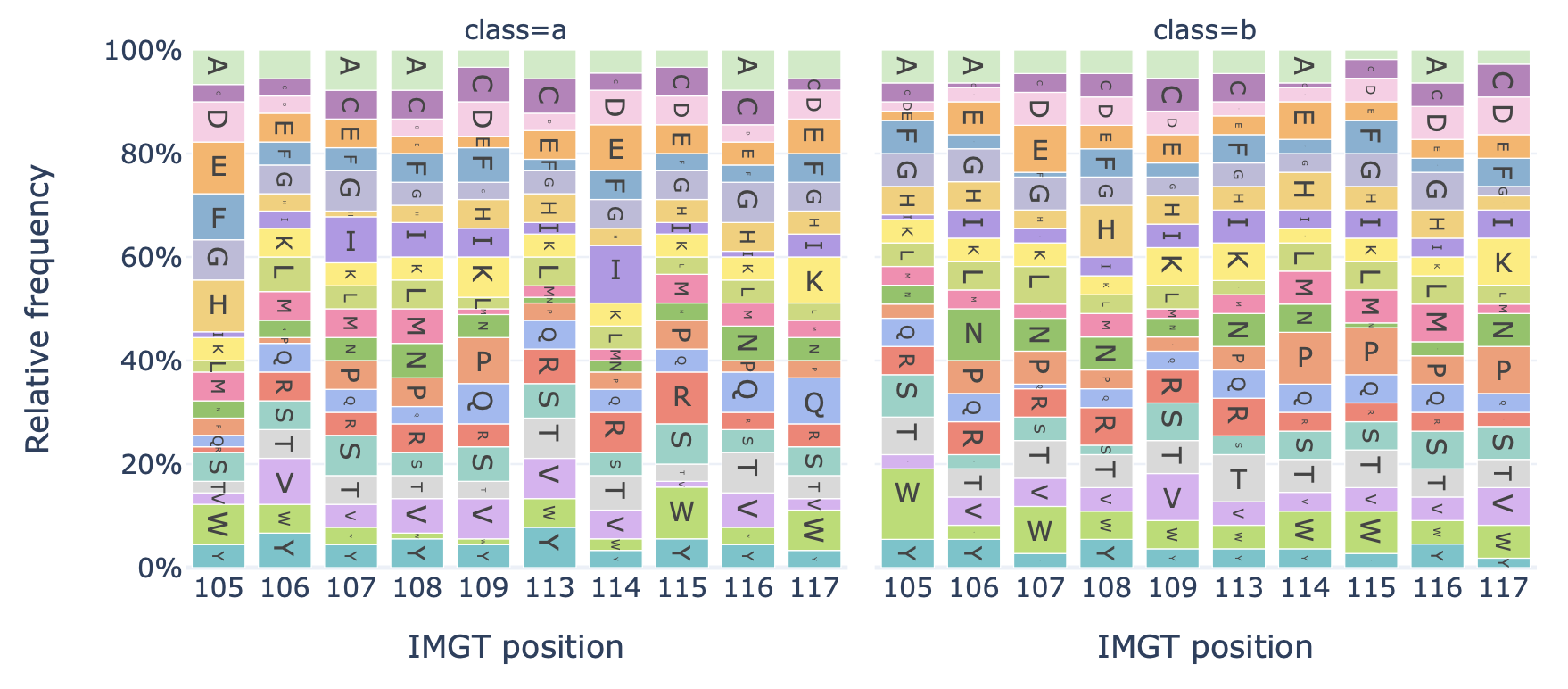
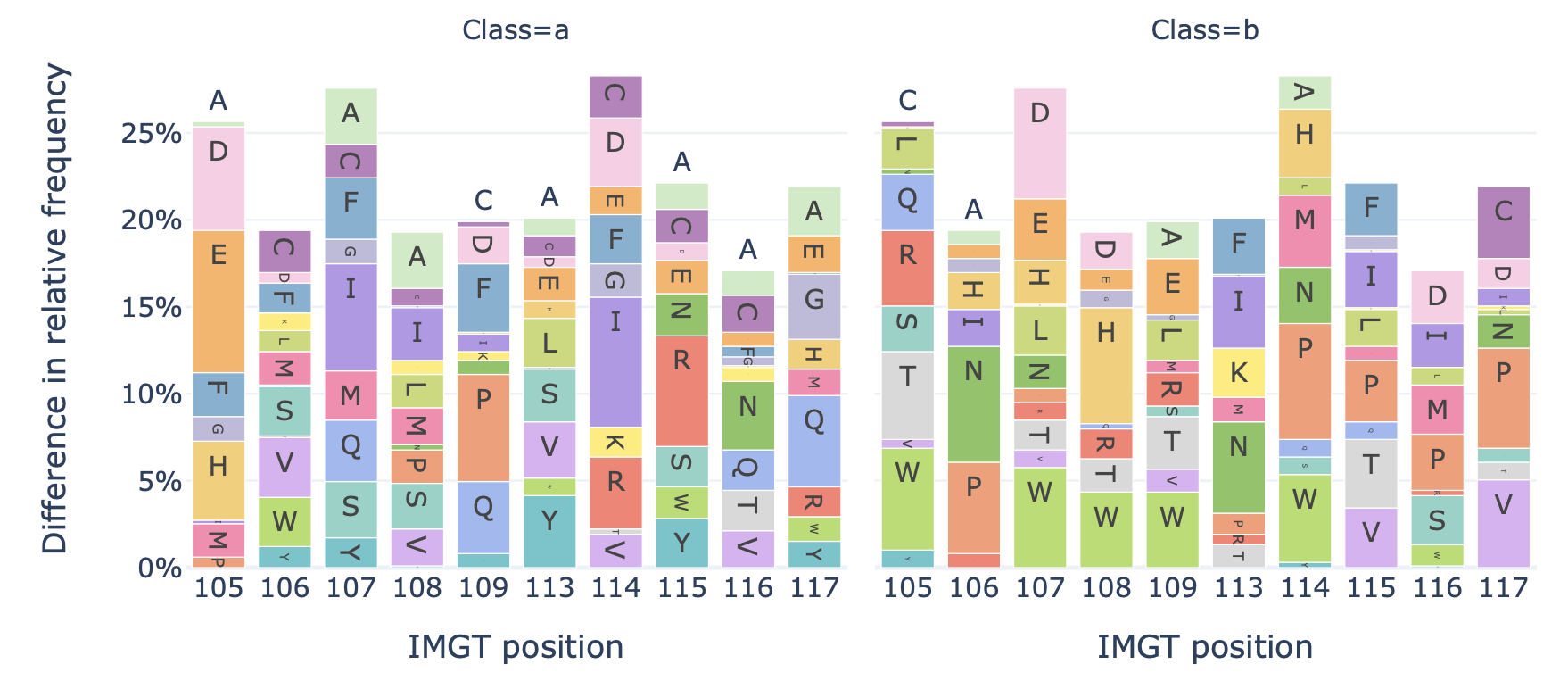
Specification arguments:
alignment (str): Alignment style for aligning sequences of different lengths. Options are as follows:
CENTER: center-align sequences of different lengths. The middle amino acid of any sequence be labelled position 0. By default, alignment is CENTER.
LEFT: left-align sequences of different lengths, starting at 0.
RIGHT: right align sequences of different lengths, ending at 0 (counting towards negative numbers).
IMGT: align sequences based on their IMGT positional numbering, considering the sequence region_type (IMGT_CDR3 or IMGT_JUNCTION). The main difference between CENTER and IMGT is that IMGT aligns the first and last amino acids, adding gaps in the middle, whereas CENTER aligns the middle of the sequences, padding with gaps at the start and end of the sequence. When region_type is IMGT_JUNCTION, the IMGT positions run from 104 (conserved C) to 118 (conserved W/F). When IMGT_CDR3 is used, these positions are 105 to 117. For long CDR3 sequences, additional numbers are added in between IMGT positions 111 and 112. See the official IMGT documentation for more details: https://www.imgt.org/IMGTScientificChart/Numbering/CDR3-IMGTgaps.html
relative_frequency (bool): Whether to plot relative frequencies (true) or absolute counts (false) of the positional amino acids. Note that when sequences are of different length, setting relative_frequency to True will produce different results depending on the alignment type, as some positions are only covered by the longest sequences. By default, relative_frequency is False.
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. If split_by_label is set to true, the percentage-wise frequency difference between classes is plotted additionally. By default, split_by_label is False.
label (str): if split_by_label is set to True, a label can be specified here.
region_type (str): which part of the sequence to check; e.g., IMGT_CDR3
YAML specification:
definitions: reports: my_aa_freq_report: AminoAcidFrequencyDistribution: relative_frequency: False split_by_label: True label: CMV region_type: IMGT_CDR3
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.CompAIRRClusteringReport module¶
- class immuneML.reports.data_reports.CompAIRRClusteringReport.CompAIRRClusteringReport(dataset: RepertoireDataset = None, result_path: Path = None, labels: List[str] = None, compairr_path: str = None, indels: bool = False, ignore_counts: bool = False, ignore_genes: bool = False, threads: int = 4, linkage_method: str = 'single', is_cdr3: bool = True, name: str = None, clustering_threshold: float = 0.5, clustering_criterion: str = 'distance')[source]¶
Bases:
DataReportA report that uses CompAIRR to compute repertoire distances based on sequence overlap and performs hierarchical clustering on the resulting distance matrix. The clustering results are visualized using a dendrogram, colored by a specified label.
The report assumes that CompAIRR (https://github.com/uio-bmi/compairr) has been installed.
Note
This is an experimental feature.
Specification arguments:
labels (list): The list of labels to highlight below the dendrogram. The labels should be present in the dataset.
compairr_path (str): Path to the CompAIRR executable.
indels (bool): Whether to allow insertions/deletions when matching sequences (default: False)
ignore_counts (bool): Whether to ignore sequence counts when computing overlap (default: False)
ignore_genes (bool): Whether to ignore V/J gene assignments when matching sequences (default: False)
threads (int): Number of threads to use for CompAIRR computation (default: 4)
linkage_method (str): The linkage method to use for hierarchical clustering (default: ‘single’)
is_cdr3 (bool): Whether the sequences represent CDR3s (default: True)
clustering_criterion (str): The criterion to use for clustering (default: ‘distance’), as defined in scipy.cluster.hierarchy.linkage; valid values are ‘distance’, ‘maxclust’, ‘monocrit’, ‘maxclust_monocrit’
clustering_threshold (float): The threshold for the clustering algorithm (default: 0.5), mapped to ‘t’ parameter in scipy.cluster.hierarchy.fcluster
YAML specification:
definitions: reports: my_compairr_clustering_report: CompAIRRClusteringReport: labels: [disease] compairr_path: /path/to/compairr indels: false ignore_counts: true ignore_genes: true threads: 4 linkage_method: single is_cdr3: true clustering_criterion: distance clustering_threshold: 0.5
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites() bool[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.DataReport module¶
- class immuneML.reports.data_reports.DataReport.DataReport(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1)[source]¶
Bases:
ReportData reports show some type of features or statistics about a given dataset.
When running the TrainMLModel instruction, data reports can be specified inside the ‘selection’ or ‘assessment’ specification under the keys ‘reports/data’ (current cross-validation split) or ‘reports/data_splits’ (train/test sub-splits). Example:
definitions: reports: my_data_report: SequenceCountDistribution my_instruction: type: TrainMLModel selection: reports: data: - my_data_report # other parameters... assessment: reports: data: - my_data_report # other parameters... # other parameters...
Alternatively, when running the ExploratoryAnalysis instruction, data reports can be specified under ‘report’. Example:
my_instruction: type: ExploratoryAnalysis analyses: my_first_analysis: report: my_data_report # other parameters... # other parameters...
- DOCS_TITLE = 'Data reports'¶
- __init__(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1)[source]¶
The arguments defined below are set at runtime by the instruction. Concrete classes inheriting DataReport may include additional parameters that will be set by the user in the form of input arguments.
dataset (Dataset): a dataset object (can be repertoire, receptor or sequence dataset, depending on the specific report) result_path (Path): location where the results (plots, tables, etc.) will be stored name (str): user-defined name of the report used in the HTML overview automatically generated by the platform number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
immuneML.reports.data_reports.GLIPH2Exporter module¶
- class immuneML.reports.data_reports.GLIPH2Exporter.GLIPH2Exporter(dataset: ReceptorDataset = None, result_path: Path = None, name: str = None, condition: str = None, number_of_processes: int = 1)[source]¶
Bases:
DataReportReport which exports the receptor data to GLIPH2 format so that it can be directly used in GLIPH2 tool. Currently, the report accepts only receptor datasets.
GLIPH2 publication: Huang H, Wang C, Rubelt F, Scriba TJ, Davis MM. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nature Biotechnology. Published online April 27, 2020:1-9. doi:10.1038/s41587-020-0505-4
Specification arguments:
condition (str): name of the parameter present in the receptor metadata in the dataset; condition can be anything which can be processed in GLIPH2, such as tissue type or treatment.
YAML specification:
definitions: reports: my_gliph2_exporter: GLIPH2Exporter: condition: epitope # for instance, epitope parameter is present in receptors' metadata with values such as "MtbLys" for Mycobacterium tuberculosis (as shown in the original paper).
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.LabelDist module¶
- class immuneML.reports.data_reports.LabelDist.LabelDist(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, labels: list = None)[source]¶
Bases:
DataReportLabelDist report plots the distribution of label values for all labels provided as input to the report.
Specification arguments:
labels (list): list of label names as they appear in the metadata file (RepertoireDataset) or in data files (Receptor/SequenceDataset).
YAML specification:
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
immuneML.reports.data_reports.LabelOverlap module¶
- class immuneML.reports.data_reports.LabelOverlap.LabelOverlap(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, column_label: str = None, row_label: str = None)[source]¶
Bases:
DataReportThis report creates a heatmap where the columns are the values of one label and rows are the values of another label, and the cells contain the number of samples that have both label values. It works for any dataset type.
Specification arguments:
column_label (str): Name of the label to be used as columns in the heatmap.
row_label (str): Name of the label to be used as rows in the heatmap.
YAML specification:
my_data_report: LabelOverlap: column_label: epitope row_label: batch
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.MotifGeneralizationAnalysis module¶
- class immuneML.reports.data_reports.MotifGeneralizationAnalysis.MotifGeneralizationAnalysis(training_set_identifier_path: str = None, training_percentage: float = None, max_positions: int = None, min_positions: int = None, min_precision: float = None, min_recall: float = None, min_true_positives: int = None, no_gaps: bool = False, test_precision_threshold: float = None, split_by_motif_size: bool = None, random_seed: int = None, label: dict = None, min_points_in_window: int = None, smoothing_constant1: float = None, smoothing_constant2: float = None, highlight_motifs_path: str = None, highlight_motifs_name: str = None, training_set_name: str = None, test_set_name: str = None, dataset: SequenceDataset = None, result_path: Path = None, number_of_processes: int = 1, name: str = None)[source]¶
Bases:
DataReportThis report splits the given dataset into a training and validation set, identifies significant motifs using the
MotifEncoderon the training set and plots the precision/recall and precision/true positive predictions of motifs on both the training and validation sets. This can be used to:determine the optimal recall cutoff for motifs of a given size
investigate how well motifs learned on a training set generalize to a test set
After running this report and determining the optimal recall cutoffs, the report
MotifTestSetPerformancecan be run to plot the performance on an independent test set.Note: the MotifEncoder (and thus this report) can only be used for sequences of the same length.
Specification arguments:
label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
training_set_identifier_path (str): Path to a file containing ‘sequence_identifiers’ of the sequences used for the training set. This file should have a single column named ‘example_id’ and have one sequence identifier per line. If training_set_identifier_path is not set, a random subset of the data (according to training_percentage) will be assigned to be the training set.
training_percentage (float): If training_set_identifier_path is not set, this value is used to specify the fraction of sequences that will be randomly assigned to form the training set. Should be a value between 0 and 1. By default, training_percentage is 0.7.
random_seed (int): Random seed for splitting the data into training and validation sets a training_set_identifier_path is not provided.
split_by_motif_size (bool): Whether to split the analysis per motif size. If true, a recall threshold is learned for each motif size, and figures are generated for each motif size independently. By default, split_by_motif_size is true.
min_precision:
MotifEncoderparameter. The minimum precision threshold for keeping a motif on the training set. By default, min_precision is 0.9.test_precision_threshold (float). The desired precision on the test set, given that motifs are learned by using a training set with a precision threshold of min_precision. It is recommended for test_precision_threshold to be lower than min_precision, e.g., min_precision - 0.1. By default, test_precision_threshold is 0.8.
min_recall (float):
MotifEncoderparameter. The minimum recall threshold for keeping a motif. Any learned recall threshold will be at least as high as the set min_recall value. The default value for min_recall is 0.min_true_positives (int):
MotifEncoderparameter. The minimum number of true positive training sequences that a motif needs to occur in. The default value for min_true_positives is 1.max_positions (int):
MotifEncoderparameter. The maximum motif size. This is number of positional amino acids the motif consists of (excluding gaps). The default value for max_positions is 4.min_positions (int):
MotifEncoderparameter. The minimum motif size (see also: max_positions). The default value for min_positions is 1.no_gaps (bool):
MotifEncoderparameter. Must be set to True if only contiguous motifs (position-specific k-mers) are allowed. By default, no_gaps is False, meaning both gapped and ungapped motifs are searched for.smoothen_combined_precision (bool): whether to add a smoothed line representing the combined precision to the precision-vs-TP plot. When set to True, this may take considerable extra time to compute. By default, plot_smoothed_combined_precision is set to True.
min_points_in_window (int): Parameter for smoothing the combined_precision line in the precision-vs-TP plot through lognormal kernel smoothing with adaptive window size. This parameter determines the minimum number of points that need to be present in a window to determine the adaptive window size. By default, min_points_in_window is 50.
smoothing_constant1: Parameter for smoothing the combined_precision line in the precision-vs-TP plot through lognormal kernel smoothing with adaptive window size. This smoothing constant determines the dependence of the smoothness on the window size. Increasing this increases smoothness for regions where few points are present. By default, smoothing_constant1 is 5.
smoothing_constant2: Parameter for smoothing the combined_precision line in the precision-vs-TP plot through lognormal kernel smoothing. with adaptive window size. This smoothing constant can be used to scale the overall kernel width, thus influencing the smoothness of all regions regardless of data density. By default, smoothing_constant2 is 10.
training_set_name (str): Name of the training set to be used in figures. By default, the training_set_name is ‘training set’.
test_set_name (str): Name of the test set to be used in figures. By default, the test_set_name is ‘test set’.
highlight_motifs_path (str): Path to a set of motifs of interest to highlight in the output figures (such as implanted ground-truth motifs). By default, no motifs are highlighted.
highlight_motifs_name (str): IF highlight_motifs_path is defined, this name will be used to label the motifs of interest in the output figures.
YAML specification:
definitions: reports: my_motif_generalization: MotifGeneralizationAnalysis: min_precision: 0.9 min_recall: 0.1 label: # Define a label, and the positive class for that given label CMV: positive_class: +
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
immuneML.reports.data_reports.NodeDegreeDistribution module¶
- class immuneML.reports.data_reports.NodeDegreeDistribution.NodeDegreeDistribution(dataset: Dataset = None, result_path: Path = None, name: str = None, compairr_path: str = None, region_type: RegionType = RegionType.IMGT_JUNCTION, indels: bool = False, ignore_genes: bool = False, hamming_distance: int = 1, per_repertoire: bool = False, per_label=False, threads: int = 4)[source]¶
Bases:
DataReportA report that uses CompAIRR to compute the node degree distribution of a sequence dataset. Results are visualized as a histogram and stored in a TSV file.
The report assumes that CompAIRR (https://github.com/uio-bmi/compairr) has been installed.
Specification arguments:
compairr_path (str): The path to the CompAIRR executable.
region_type (str): The region type to analyze. Can be either “IMGT_CDR3” or “IMGT_JUNCTION”.
indels (bool): Whether to include indels in the analysis. Default is False.
ignore_genes (bool): Whether to ignore gene names in the analysis. Default is False.
hamming_distance (int): The Hamming distance to use for the analysis. Default is 1.
per_repertoire (bool): Whether to compute the node degree distribution for each repertoire separately. Only applicable when using a RepertoireDataset. Default is False.
per_label (bool): Whether to compute the node degree distribution for each label separately. Only applicable when using a RepertoireDataset. Default is False.
threads (int): The number of threads to use for the analysis. Default is 4.
YAML specification:
NodeDegreeDistribution: compairr_path: /path/to/compairr region_type: IMGT_JUNCTION indels: False ignore_genes: False hamming_distance: 1 per_repertoire: False per_label: False threads: 4
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites() bool[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.RecoveredSignificantFeatures module¶
- class immuneML.reports.data_reports.RecoveredSignificantFeatures.RecoveredSignificantFeatures(dataset: RepertoireDataset = None, ground_truth_sequences_path: Path = None, p_values: List[float] = None, k_values: List[int] = None, label: dict = None, compairr_path: Path = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, region_type: RegionType = None, sequence_type: SequenceType = None)[source]¶
Bases:
DataReportCompares a given collection of ground truth implanted signals (sequences or k-mers) to the significant label-associated k-mers or sequences according to Fisher’s exact test.
Internally uses the
KmerAbundanceEncoderfor calculating significant k-mers, andSequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoderto calculate significant full sequences (depending on whether the argument compairr_path was set).This report creates two plots:
the first plot is a bar chart showing what percentage of the ground truth implanted signals were found to be significant.
the second plot is a bar chart showing what percentage of the k-mers/sequences found to be significant match the ground truth implanted signals.
To compare k-mers or sequences of differing lengths, the ground truth sequences or long k-mers are split into k-mers of the given size through a sliding window approach. When comparing ‘full_sequences’ to ground truth sequences, a match is only registered if both sequences are of equal length.
Specification arguments:
ground_truth_sequences_path (str): Path to a file containing the true implanted (sub)sequences, e.g., full sequences or k-mers. The file should contain one sequence per line, without a header, and without V or J genes.
sequence_type (str): either amino acid or nucleotide; which type of sequence to use for the analysis
region_type (str): which AIRR field to use for comparison, e.g. IMGT_CDR3 or IMGT_JUNCTION
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. When using a full sequence encoding (SequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoder), specify ‘full_sequence’ here. Each value specified under k_values will represent one bar in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
compairr_path (str): If ‘full_sequence’ is listed under k_values, the path to the CompAIRR executable may be provided. If the compairr_path is specified, the
CompAIRRSequenceAbundanceEncoderwill be used to compute the significant sequences. If the path is not specified and ‘full_sequence’ is listed under k-values,SequenceAbundanceEncoderwill be used.
YAML specification:
definitions: reports: my_recovered_significant_features_report: RecoveredSignificantFeatures: groundtruth_sequences_path: path/to/groundtruth/sequences.txt trim_leading_trailing: False p_values: - 0.1 - 0.01 - 0.001 - 0.0001 k_values: - 3 - 4 - 5 - full_sequence compairr_path: path/to/compairr # can be specified if 'full_sequence' is listed under k_values label: # Define a label, and the positive class for that given label CMV: positive_class: +
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.RepertoireClonotypeSummary module¶
- class immuneML.reports.data_reports.RepertoireClonotypeSummary.RepertoireClonotypeSummary(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, color_label: str = None, facet_label: str = None)[source]¶
Bases:
DataReportShows the number of distinct clonotypes per repertoire in a given dataset as a bar plot.
Specification arguments:
color_label (str): the label to color the bar plot by (optional, default: None)
facet_label (str): the label to facet the bar plot by (optional, default: None)
YAML specification:
definitions: reports: my_clonotype_summary_rep: RepertoireClonotypeSummary: color_label: celiac facet_label: hla
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites() bool[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.SequenceCountDistribution module¶
- class immuneML.reports.data_reports.SequenceCountDistribution.SequenceCountDistribution(dataset: Dataset = None, result_path: Path = None, number_of_processes: int = 1, split_by_label: bool = None, label: str = None, name: str = None)[source]¶
Bases:
DataReportGenerates a histogram of the duplicate counts of the sequences in a dataset.
Specification arguments:
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. By default, split_by_label is False.
label (str): Optional label for separating the results by color/creating separate plots. Note that this should the name of a valid dataset label.
YAML specification:
my_sld_report: SequenceCountDistribution: label: disease
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.SequenceLengthDistribution module¶
- class immuneML.reports.data_reports.SequenceLengthDistribution.SequenceLengthDistribution(dataset: Dataset = None, batch_size: int = 1, result_path: Path = None, number_of_processes: int = 1, region_type: RegionType = RegionType.IMGT_CDR3, sequence_type: SequenceType = SequenceType.AMINO_ACID, name: str = None, label: str = None, split_by_label: bool = False, plot_frequencies: bool = False)[source]¶
Bases:
DataReportGenerates a histogram of the lengths of the sequences in a dataset.
Specification arguments:
sequence_type (str): whether to check the length of amino acid or nucleotide sequences; default value is ‘amino_acid’
region_type (str): which part of the sequence to examine; e.g., IMGT_CDR3
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. By default, split_by_label is False.
label (str): if split_by_label is set to True, a label can be specified here.
plot_frequencies (bool): if set to True, the plot will show the frequencies of the sequence lengths instead of the counts. By default, plot_frequencies is False.
YAML specification:
definitions: reports: my_sld_report: SequenceLengthDistribution: sequence_type: amino_acid region_type: IMGT_CDR3 label: label_1 split_by_label: True plot_frequencies: True
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.SequencesWithSignificantKmers module¶
- class immuneML.reports.data_reports.SequencesWithSignificantKmers.SequencesWithSignificantKmers(dataset: RepertoireDataset = None, reference_sequences_path: Path = None, p_values: List[float] = None, k_values: List[int] = None, label: dict = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, sequence_type: SequenceType = None, region_type: RegionType = None)[source]¶
Bases:
DataReportGiven a list of reference sequences, this report writes out the subsets of reference sequences containing significant k-mers (as computed by the
KmerAbundanceEncoderusing Fisher’s exact test).For each combination of p-value and k-mer size given, a file is written containing all sequences containing a significant k-mer of the given size at the given p-value.
Specification arguments:
reference_sequences_path (str): Path to a file containing the reference sequences, The file should contain one sequence per line, without a header, and without V or J genes.
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. Each k-mer length will become one panel in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
YAML specification:
definitions: reports: my_sequences_with_significant_kmers: SequencesWithSignificantKmers: reference_sequences_path: path/to/reference/sequences.txt p_values: - 0.1 - 0.01 - 0.001 - 0.0001 k_values: - 3 - 4 - 5 label: # Define a label, and the positive class for that given label CMV: positive_class: +
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.ShannonDiversityOverview module¶
- class immuneML.reports.data_reports.ShannonDiversityOverview.ShannonDiversityOverview(dataset: RepertoireDataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, color_label: str = None, facet_row_label: str = None, facet_col_label: str = None)[source]¶
Bases:
DataReportComputes Shannon diversity for each repertoire using Shannon diversity encoder and plots the results in a histogram, optionally stratified by labels.
Dataset type:
Repertoire Dataset
Specification arguments:
color_label (str): The label used to color the histogram bars. Default is None.
facet_row_label (str): The label used to facet the histogram into multiple rows. Default is None, meaning no row faceting.
facet_col_label (str): The label used to facet the histogram into multiple columns. Default is None, meaning no column faceting.
YAML specification:
definitions: reports: shannon_div_rep: ShannonDiversityOverview: color_label: disease_status
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites() bool[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
- prepare_data(encoded_dataset) Tuple[DataFrame, ReportOutput][source]¶
immuneML.reports.data_reports.SignificantFeatures module¶
- class immuneML.reports.data_reports.SignificantFeatures.SignificantFeatures(dataset: RepertoireDataset = None, p_values: List[float] = None, k_values: List[int] = None, label: dict = None, compairr_path: Path = None, log_scale: bool = False, result_path: Path = None, name: str = None, number_of_processes: int = 1, region_type: RegionType = None, sequence_type: SequenceType = None)[source]¶
Bases:
DataReportPlots a boxplot of the number of significant features (label-associated k-mers or sequences) per Repertoire according to Fisher’s exact test, across different classes for the given label.
Internally uses the
KmerAbundanceEncoderfor calculating significant k-mers, andSequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoderto calculate significant full sequences (depending on whether the argument compairr_path was set).Specification arguments:
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. When using a full sequence encoding (SequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoder), specify ‘full_sequence’ here. Each value specified under k_values will represent one boxplot in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
compairr_path (str): If ‘full_sequence’ is listed under k_values, the path to the CompAIRR executable may be provided. If the compairr_path is specified, the
CompAIRRSequenceAbundanceEncoderwill be used to compute the significant sequences. If the path is not specified and ‘full_sequence’ is listed under k-values,SequenceAbundanceEncoderwill be used.log_scale (bool): Whether to plot the y axis in log10 scale (log_scale = True) or continuous scale (log_scale = False). By default, log_scale is False.
YAML specification:
definitions: reports: my_significant_features_report: SignificantFeatures: p_values: - 0.1 - 0.01 - 0.001 - 0.0001 k_values: - 3 - 4 - 5 - full_sequence compairr_path: path/to/compairr # can be specified if 'full_sequence' is listed under k_values label: # Define a label, and the positive class for that given label CMV: positive_class: + log_scale: False
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.SignificantKmerPositions module¶
- class immuneML.reports.data_reports.SignificantKmerPositions.SignificantKmerPositions(dataset: RepertoireDataset = None, reference_sequences_path: Path = None, p_values: List[float] = None, k_values: List[int] = None, label: dict = None, compairr_path: Path = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, region_type: RegionType = None, sequence_type: SequenceType = None)[source]¶
Bases:
DataReportPlots the number of significant k-mers (as computed by the
KmerAbundanceEncoderusing Fisher’s exact test) observed at each IMGT position of a given list of reference sequences. This report creates a stacked bar chart, where each bar represents an IMGT position, and each segment of the stack represents the observed frequency of one ‘significant’ k-mer at that position.Specification arguments:
reference_sequences_path (str): Path to a file containing the reference sequences, The file should contain one sequence per line, without a header, and without V or J genes.
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. Each k-mer length will become one panel in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
sequence_type (str): nucleotide or amino_acid
region_type (str): which AIRR field to consider, e.g., IMGT_CDR3 or IMGT_JUNCTION
YAML specification:
definitions: reports: my_significant_kmer_positions_report: SignificantKmerPositions: reference_sequences_path: path/to/reference/sequences.txt p_values: - 0.1 - 0.01 - 0.001 - 0.0001 k_values: - 3 - 4 - 5 label: # Define a label, and the positive class for that given label CMV: positive_class: +
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.TrueMotifsSummaryBarplot module¶
- class immuneML.reports.data_reports.TrueMotifsSummaryBarplot.TrueMotifsSummaryBarplot(implanted_motifs_per_signal, dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, region_type: RegionType = RegionType.IMGT_CDR3)[source]¶
Bases:
DataReportThis report can be used to show how well motifs (for example, motifs introduced using the Simulation instruction) are learned across different generative models. The report shows a bar plot with the proportion of sequences in each dataset that contain the given motifs. Bars are grouped by the dataset origin (e.g., train, PWM, VAE, LSTM) and the signals provided. The report also shows how many of the sequences are memorized (seen in train data) and how many are novel (not seen in train data).
Specification arguments:
region_type (str): which part of the sequence to check; e.g., IMGT_CDR3
implanted_motifs_per_signal (dict): a nested dictionary that specifies the motif seeds that were implanted in the given dataset. The first level of keys in this dictionary represents the different signals. In the inner dictionary there should be two keys: “seeds” and “gap_sizes”.
seeds: a list of motif seeds. The seeds may contain gaps, specified by a ‘/’ symbol.
gap_sizes: a list of all the possible gap sizes that were used when implanting a gapped motif seed. When no gapped seeds are used, this value has no effect.
YAML specification:
definitions: reports: my_motif_report: TrueMotifsSummaryBarplot: region_type: IMGT_CDR3 implanted_motifs_per_signal: my_signal1: seeds: - DEQ gap_sizes: - 0 my_signal2: seeds: - AS/G gap_sizes: - 2
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.data_reports.VJGeneDistribution module¶
- class immuneML.reports.data_reports.VJGeneDistribution.VJGeneDistribution(dataset: Dataset = None, result_path: Path = None, number_of_processes: int = 1, name: str = None, split_by_label: bool = None, label: str = None, is_sequence_label: bool = None, show_joint_dist: bool = True)[source]¶
Bases:
DataReportThis report creates several plots to gain insight into the V and J gene distribution of a given dataset. When a label is provided, the information in the plots is separated per label value, either by color or by creating separate plots. This way one can for example see if a particular V or J gene is more prevalent across disease associated receptors.
Individual V and J gene distributions: for sequence and receptor datasets, a bar plot is created showing how often each V or J gene occurs in the dataset. For repertoire datasets, boxplots are used to represent how often each V or J gene is used across all repertoires. Since repertoires may differ in size, these counts are normalised by the repertoire size (original count values are additionaly exported in tsv files).
Combined V and J gene distributions: for sequence and receptor datasets, a heatmap is created showing how often each combination of V and J genes occurs in the dataset. A similar plot is created for repertoire datasets, except in this case only the average value for the normalised gene usage frequencies are shown (original count values are additionaly exported in tsv files).
Specification arguments:
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. By default, split_by_label is False.
label (str): Optional label for separating the results by color/creating separate plots. Note that this should the name of a valid dataset label.
is_sequence_label (bool): for RepertoireDatasets, indicates if the label applies to the sequence level (e.g., antigen binding versus non-binding across repertoires) or repertoire level (e.g., diseased repertoires versus healthy repertoires). By default, is_sequence_label is False. For Sequence- and ReceptorDatasets, this parameter is ignored.
show_joint_dist (bool): whether to show the combined V and J gene distribution. Default is True.
YAML specification:
definitions: reports: my_vj_gene_report: VJGeneDistribution: label: ag_binding show_joint_dist: false
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.