immuneML.reports.encoding_reports package¶
Submodules¶
immuneML.reports.encoding_reports.DesignMatrixExporter module¶
- class immuneML.reports.encoding_reports.DesignMatrixExporter.DesignMatrixExporter(dataset: Dataset = None, result_path: Path = None, file_format: str = None, number_of_processes: int = 1, name: str = None)[source]¶
Bases:
EncodingReportExports the design matrix and related information of a given encoded Dataset to csv files. If the encoded data has more than 2 dimensions (such as when using the OneHot encoder with option Flatten=False), the data are then exported to different formats to facilitate their import with external software.
Specification arguments:
file_format (str): the format and extension of the file to store the design matrix. The supported formats are: npy, csv, pt, hdf5, npy.zip, csv.zip or hdf5.zip.
Note: when using hdf5 or hdf5.zip output formats, make sure the ‘hdf5’ dependency is installed.
YAML specification:
definitions: reports: my_dme_report: DesignMatrixExporter: file_format: csv
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.DimensionalityReduction module¶
- class immuneML.reports.encoding_reports.DimensionalityReduction.DimensionalityReduction(dataset: Dataset = None, batch_size: int = 1, result_path: Path = None, name: str = None, labels: list = None, dim_red_method: DimRedMethod = None, components: list = None)[source]¶
Bases:
EncodingReportThis report visualizes the data obtained by dimensionality reduction. The data points can be highlighted by label of interest. It is also possible to specify labels that contain lists of values (e.g., HLA), in which case the data points will be duplicated (so that each point refers to one HLA allele) and jittered slightly to improve visibility before being highlighted by the concrete HLA allele values.
When a
dim_red_methodis configured, itscomponentsparameter determines which two components are plotted and overrides the report-levelcomponents. When nodim_red_methodis set (using pre-computed dimensionality-reduced data), the report-levelcomponentsselects which two columns to plot. All computed components are always written to the output CSV.For PCA the explained variance per component is exported to a separate CSV and annotated on the axis labels. For KernelPCA with
compute_total_variance: truethe fraction of total kernel-space variance is shown instead.Specification arguments:
labels (list): names of the label to use for highlighting data points; or None
components (list): which two components (1-indexed) to plot when no
dim_red_methodis provided. When adim_red_methodis set, use its owncomponentsparameter instead. Default: [1, 2].dim_red_method (str): dimensionality reduction method to be used for plotting; if set, in a workflow, this dimensionality reduction will be used for plotting instead of any other set in the workflow; if None, it will visualize the encoded data of reduced dimensionality if set
YAML specification:
definitions: reports: # Plot PC3 vs PC4 from a 5-component PCA, annotated with explained variance rep1: DimensionalityReduction: labels: [epitope, source] dim_red_method: PCA: n_components: 5 components: [3, 4] # Plot components 1 vs 2 from pre-computed dimensionality-reduced data rep2: DimensionalityReduction: labels: [epitope] components: [1, 2]
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.EncodingReport module¶
- class immuneML.reports.encoding_reports.EncodingReport.EncodingReport(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1)[source]¶
Bases:
ReportEncoding reports show some type of features or statistics about an encoded dataset, or may in some cases export relevant sequences or tables.
When running the TrainMLModel instruction, encoding reports can be specified inside the ‘selection’ or ‘assessment’ specification under the key ‘reports/encoding’. Example:
my_instruction: type: TrainMLModel selection: reports: encoding: - my_encoding_report # other parameters... assessment: reports: encoding: - my_encoding_report # other parameters... # other parameters...
Alternatively, when running the ExploratoryAnalysis instruction, encoding reports can be specified under ‘report’. Example:
my_instruction: type: ExploratoryAnalysis analyses: my_first_analysis: report: my_encoding_report # other parameters... # other parameters...
- DOCS_TITLE = 'Encoding reports'¶
- __init__(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1)[source]¶
The arguments defined below are set at runtime by the instruction. Concrete classes inheriting EncodingReport may include additional parameters that will be set by the user in the form of input arguments.
dataset (Dataset): an encoded dataset where encoded_data attribute is set to an instance of EncodedData object result_path (Path): path where the results will be stored (plots, tables, etc.) name (str): user-defined name of the report that will be shown in the HTML overview later number_of_processes (int): how many processes should be created at once to speed up the analysis. For personal machines, 4 or 8 is usually a good choice.
immuneML.reports.encoding_reports.FeatureComparison module¶
- class immuneML.reports.encoding_reports.FeatureComparison.FeatureComparison(dataset: Dataset = None, result_path: Path = None, comparison_label: str = None, color_grouping_label: str = None, row_grouping_label=None, column_grouping_label=None, opacity: float = 0.7, error_function: str = 'std', name: str = None, show_error_bar=True, log_scale: bool = False, keep_fraction: int = 1, number_of_processes: int = 1)[source]¶
Bases:
FeatureReportEncoding a dataset results in a numeric matrix, where the rows are examples (e.g., sequences, receptors, repertoires) and the columns are features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
This report separates the examples based on a binary metadata label, and plots the mean feature value of each feature in one example group against the other example group (for example: plot the feature value of ‘sick’ repertoires on the x axis, and ‘healthy’ repertoires on the y axis to spot consistent differences). The plot can be separated into different colors or facets using other metadata labels (for example: plot the average feature values of ‘cohort1’, ‘cohort2’ and ‘cohort3’ in different colors to spot biases).
Alternatively, when plotting features without comparing them across a binary label, see:
FeatureValueBarplotreport to plot a simple bar chart per feature (average across examples). OrFeatureDistributionreport to plot the distribution of each feature across examples, rather than only showing the mean value in a bar plot.Example output:
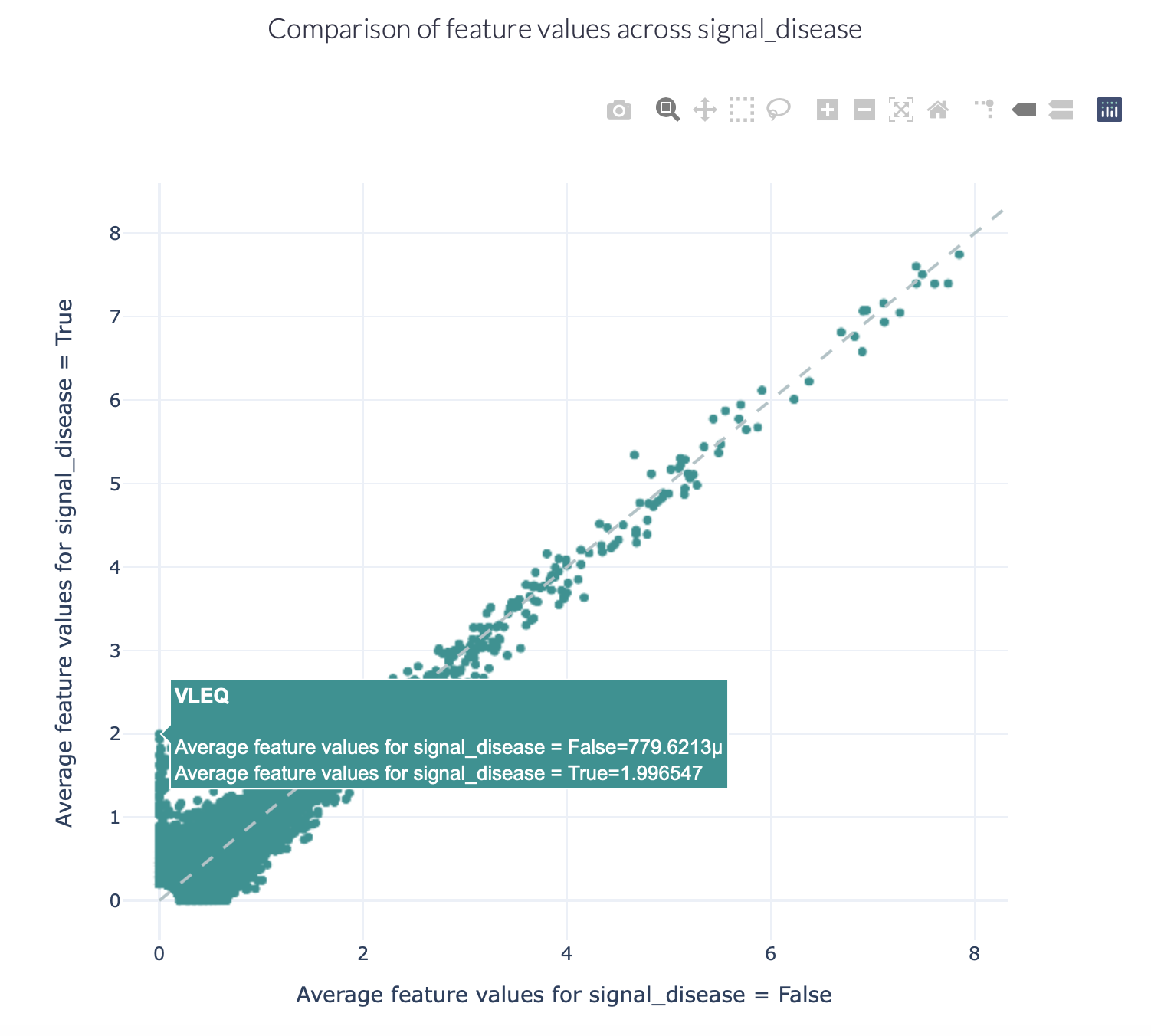
Specification arguments:
comparison_label (str): Mandatory label. This label is used to split the encoded data matrix and define the x and y axes of the plot. This label is only allowed to have 2 classes (for example: sick and healthy, binding and non-binding).
color_grouping_label (str): Optional label that is used to color the points in the scatterplot. This can not be the same as comparison_label.
row_grouping_label (str): Optional label that is used to group scatterplots into different row facets. This can not be the same as comparison_label.
column_grouping_label (str): Optional label that is used to group scatterplots into different column facets. This can not be the same as comparison_label.
show_error_bar (bool): Whether to show the error bar (standard deviation) for the points, both in the x and y dimension.
log_scale (bool): Whether to plot the x and y axes in log10 scale (log_scale = True) or continuous scale (log_scale = False). By default, log_scale is False.
keep_fraction (float): The total number of features may be very large and only the features differing significantly across comparison labels may be of interest. When the keep_fraction parameter is set below 1, only the fraction of features that differs the most across comparison labels is kept for plotting (note that the produced .csv file still contains all data). By default, keep_fraction is 1, meaning that all features are plotted.
opacity (float): a value between 0 and 1 setting the opacity for data points making it easier to see if there are overlapping points
error_function (str): which error function to use for the error bar. Options are ‘std’ (standard deviation) or ‘sem’ (standard error of the mean). Default: std.
YAML specification:
definitions: reports: my_comparison_report: FeatureComparison: # compare the different classes defined in the label disease comparison_label: disease
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.FeatureDistribution module¶
- class immuneML.reports.encoding_reports.FeatureDistribution.FeatureDistribution(dataset: Dataset = None, result_path: Path = None, color_grouping_label: str = None, row_grouping_label=None, column_grouping_label=None, mode: str = 'auto', x_title: str = None, y_title: str = None, number_of_processes: int = 1, name: str = None, error_function: str = None, plot_top_n: int = None, plot_bottom_n: int = None, plot_all_features: bool = True)[source]¶
Bases:
FeatureReportEncoding a dataset results in a numeric matrix, where the rows are examples (e.g., sequences, receptors, repertoires) and the columns are features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
This report plots the distribution of feature values. For each feature, a violin plot is created to show the distribution of feature values across all examples. The violin plots can be separated into different colors or facets using metadata labels (for example: plot the feature distributions of ‘cohort1’, ‘cohort2’ and ‘cohort3’ in different colors to spot biases).
See also:
FeatureValueBarplotreport to plot a simple bar chart per feature (average across examples), rather than the entire distribution. OrFeatureComparisonreport to compare features across binary metadata labels (e.g., plot the feature value of ‘sick’ repertoires on the x axis, and ‘healthy’ repertoires on the y axis).Example output:
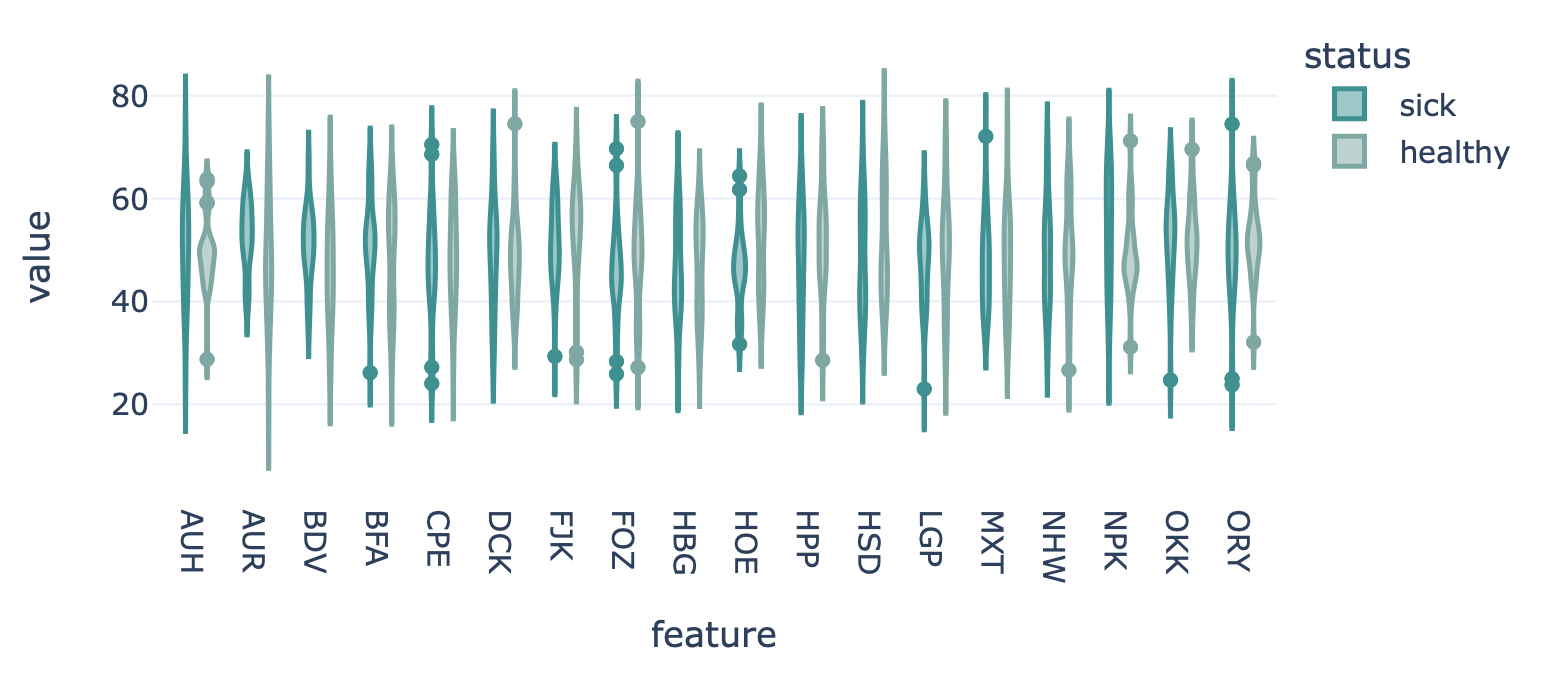
Specification arguments:
color_grouping_label (str): The label that is used to color each bar, at each level of the grouping_label.
row_grouping_label (str): The label that is used to group bars into different row facets.
column_grouping_label (str): The label that is used to group bars into different column facets.
mode (str): either ‘normal’, ‘sparse’ or ‘auto’ (default). in the ‘normal’ mode there are normal boxplots corresponding to each column of the encoded dataset matrix; in the ‘sparse’ mode all zero cells are eliminated before passing the data to the boxplots. If mode is set to ‘auto’, then it will automatically set to ‘sparse’ if the density of the matrix is below 0.01
x_title (str): x-axis label
y_title (str): y-axis label
plot_top_n (int): plot n of the largest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels.
plot_bottom_n (int): plot n of the smallest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels.
plot_all_features (bool): whether to plot all (might be slow for large number of features)
error_function (str): which error function to use for the error bar. Options are ‘std’ (standard deviation) or ‘sem’ (standard error of the mean). Default: std.
YAML specification:
definitions: reports: my_fdistr_report: FeatureDistribution: mode: sparse plot_all_features: True plot_top_n: 10 plot_bottom_n: 10
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
immuneML.reports.encoding_reports.FeatureReport module¶
- class immuneML.reports.encoding_reports.FeatureReport.FeatureReport(dataset: Dataset = None, result_path: Path = None, color_grouping_label: str = None, row_grouping_label=None, column_grouping_label=None, name: str = None, number_of_processes: int = 1, error_function: str = None)[source]¶
Bases:
EncodingReportBase class for reports that plot something about the reshaped feature values of any dataset.
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.FeatureValueBarplot module¶
- class immuneML.reports.encoding_reports.FeatureValueBarplot.FeatureValueBarplot(dataset: RepertoireDataset = None, result_path: Path = None, color_grouping_label: str = None, row_grouping_label=None, column_grouping_label=None, x_title: str = None, y_title: str = None, show_error_bar=True, name: str = None, plot_all_features: bool = True, error_function: str = None, number_of_processes: int = 1, plot_top_n: int = None, plot_bottom_n: int = None)[source]¶
Bases:
FeatureReportEncoding a dataset results in a numeric matrix, where the rows are examples (e.g., sequences, receptors, repertoires) and the columns are features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
This report plots the mean feature values per feature. A bar plot is created where the average feature value across all examples is shown, with optional error bars. The bar plots can be separated into different colors or facets using metadata labels (for example: plot the average feature values of ‘cohort1’, ‘cohort2’ and ‘cohort3’ in different colors to spot biases).
See also:
FeatureDistributionreport to plot the distribution of each feature across examples, rather than only showin the mean value in a bar plot. OrFeatureComparisonreport to compare features across binary metadata labels (e.g., plot the feature value of ‘sick’ repertoires on the x axis, and ‘healthy’ repertoires on the y axis.).Example output:
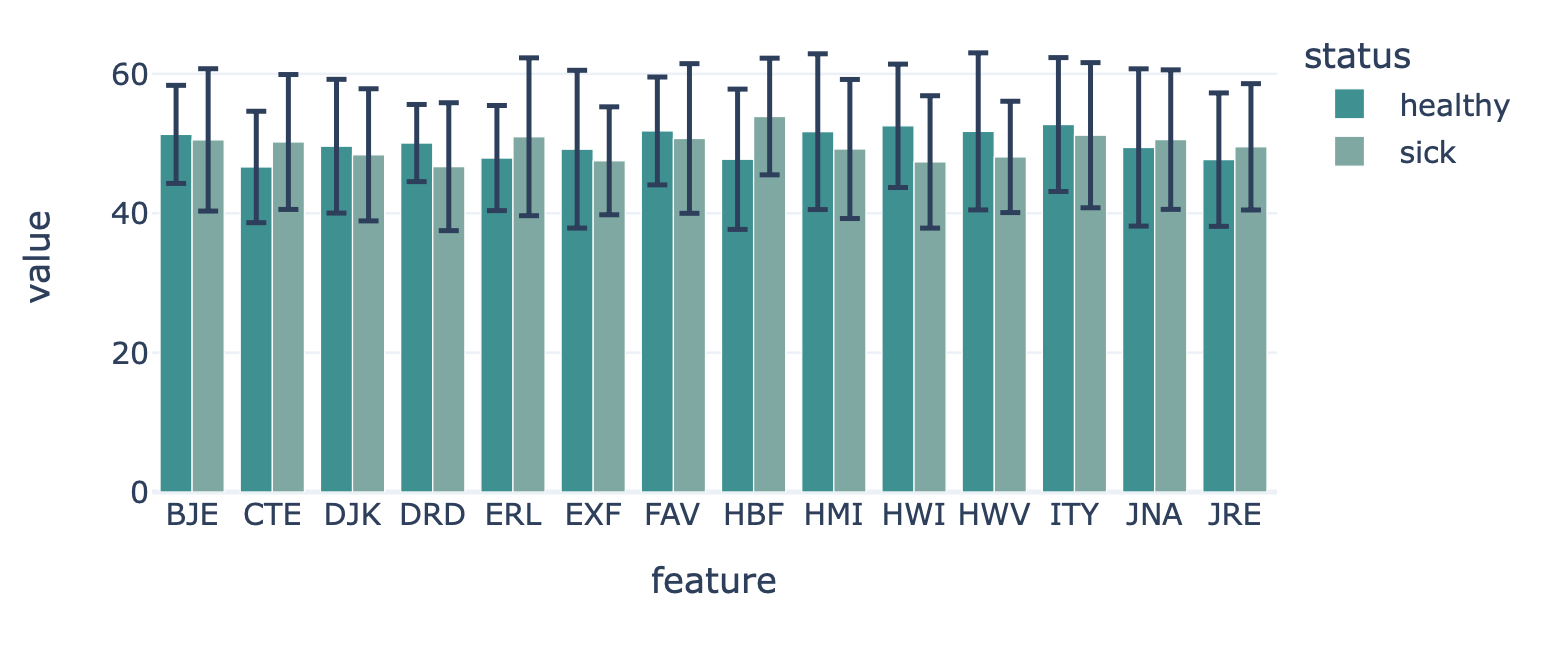
Specification arguments:
color_grouping_label (str): The label that is used to color each bar, at each level of the grouping_label.
row_grouping_label (str): The label that is used to group bars into different row facets.
column_grouping_label (str): The label that is used to group bars into different column facets.
show_error_bar (bool): Whether to show the error bar (standard deviation) for the bars.
x_title (str): x-axis label
y_title (str): y-axis label
plot_top_n (int): plot n of the largest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels. The plot shows averages per label classes.
plot_bottom_n (int): plot n of the smallest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels. The plot shows averages per label classes.
plot_all_features (bool): whether to plot all (might be slow for large number of features)
error_function (str): which error function to use for the error bar. Options are ‘std’ (standard deviation) or ‘sem’ (standard error of the mean). Default: std.
YAML specification:
definitions: reports: my_fvb_report: FeatureValueBarplot: # timepoint, disease_status and age_group are metadata labels column_grouping_label: timepoint row_grouping_label: disease_status color_grouping_label: age_group plot_all_features: true plot_top_n: 10 plot_bottom_n: 5
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
immuneML.reports.encoding_reports.GroundTruthMotifOverlap module¶
- class immuneML.reports.encoding_reports.GroundTruthMotifOverlap.GroundTruthMotifOverlap(dataset: Dataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1, groundtruth_motifs_path: str = None)[source]¶
Bases:
EncodingReportCreates report displaying overlap between learned motifs and groundtruth motifs implanted in a given sequence dataset. This report must be used in combination with the MotifEncoder.
Specification arguments:
groundtruth_motifs_path (str): Path to a .tsv file containing groundtruth position-specific motifs. The file should specify the motifs as position-specific amino acids, one column representing the positions concatenated with an ‘&’ symbol, the next column specifying the amino acids concatenated with ‘&’ symbol, and the last column specifying the implant rate.
Example:
indices
amino_acids
n_sequences
0
A
4
4&8&9
G&A&C
30
This file shows a motif ‘A’ at position 0 implanted in 4 sequences, and motif G—AC implanted between positions 4 and 9 in 30 sequences
YAML specification:
definitions: reports: my_ground_truth_motif_report: GroundTruthMotifOverlap: groundtruth_motifs_path: path/to/file.tsv
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.Matches module¶
- class immuneML.reports.encoding_reports.Matches.Matches(dataset: RepertoireDataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1)[source]¶
Bases:
EncodingReportReports the number of matches that were found when using one of the following encoders:
MatchedSequences encoder
MatchedReceptors encoder
MatchedRegex encoder
Report results are:
A table containing all matches, where the rows correspond to the Repertoires, and the columns correspond to the objects to match (regular expressions or receptor sequences).
The repertoire sizes (read frequencies and the number of unique sequences per repertoire), for each of the chains. This can be used to calculate the percentage of matched sequences in a repertoire.
When using MatchedSequences encoder or MatchedReceptors encoder, tables describing the chains and receptors (ids, chains, V and J genes and sequences).
When using MatchedReceptors encoder or using MatchedRegex encoder with chain pairs, tables describing the paired matches (where a match was found in both chains) per repertoire.
YAML specification:
definitions: reports: my_match_report: Matches
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.MotifTestSetPerformance module¶
- class immuneML.reports.encoding_reports.MotifTestSetPerformance.MotifTestSetPerformance(dataset: Dataset = None, result_path: Path = None, test_dataset_import_cls: Type[DataImport] = None, test_dataset_import_params: DatasetImportParams = None, training_set_name: str = None, test_set_name: str = None, split_by_motif_size: bool = None, highlight_motifs_path: str = None, highlight_motifs_name: str = None, min_points_in_window: int = None, smoothing_constant1: float = None, smoothing_constant2: float = None, keep_test_dataset: bool = None, number_of_processes: int = 1, name: str = None)[source]¶
Bases:
EncodingReportThis report can be used to show the performance of a learned set motifs using the
MotifEncoderon an independent test set of unseen data.It is recommended to first run the report
MotifGeneralizationAnalysisin order to calibrate the optimal recall thresholds and plot the performance of motifs on training- and validation sets.Specification arguments:
test_dataset (dict): parameters for importing a SequenceDataset to use as an independent test set. By default, the import parameters ‘is_repertoire’ and ‘paired’ will be set to False to ensure a SequenceDataset is imported.
YAML specification:
definitions: reports: my_motif_report: MotifTestSetPerformance: test_dataset: format: AIRR # choose any valid import format params: path: path/to/files/ is_repertoire: False # is_repertoire must be False to import a SequenceDataset paired: False # paired must be False to import a SequenceDataset # optional other parameters...
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites() bool[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.NonMotifSequenceSimilarity module¶
- class immuneML.reports.encoding_reports.NonMotifSequenceSimilarity.NonMotifSequenceSimilarity(dataset: SequenceDataset = None, result_path: Path = None, motif_color_map: dict = None, name: str = None, number_of_processes: int = 1)[source]¶
Bases:
EncodingReportPlots the similarity of positions outside the motifs of interest. This report can be used to investigate if the motifs of interest as determined by the
MotifEncoderhave a tendency occur in sequences that are naturally very similar or dissimilar.For each motif, the subset of sequences containing the motif is selected, and the hamming distances are computed between all sequences in this subset. Finally, a plot is created showing the distribution of hamming distances between the sequences containing the motif. For motifs occurring in sets of very similar sequences, this distribution will lean towards small hamming distances. Likewise, for motifs occurring in a very diverse set of sequences, the distribution will lean towards containing more large hamming distances.
Specification arguments:
motif_color_map (dict): An optional mapping between motif sizes and colors. If no mapping is given, default colors will be chosen.
YAML specification:
definitions: reports: my_motif_sim: NonMotifSimilarity: motif_color_map: 3: "#66C5CC" 4: "#F6CF71" 5: "#F89C74"
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.PositionalMotifFrequencies module¶
- class immuneML.reports.encoding_reports.PositionalMotifFrequencies.PositionalMotifFrequencies(dataset: SequenceDataset = None, result_path: Path = None, motif_color_map: dict = None, name: str = None, number_of_processes: int = 1)[source]¶
Bases:
EncodingReportThis report must be used in combination with the
MotifEncoder. Plots a stacked bar plot of amino acid occurrence at different indices in any given dataset, along with a plot investigating motif continuity which displays a bar plot of the gap sizes between the amino acids in the motifs in the given dataset. Note that a distance of 1 means that the amino acids are continuous (next to each other).Specification arguments:
motif_color_map (dict): Optional mapping between motif lengths and specific colors to be used. Example:
- motif_color_map:
1: #66C5CC 2: #F6CF71 3: #F89C74
YAML specification:
definitions: reports: my_pos_motif_report: PositionalMotifFrequencies: motif_color_map:
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.
immuneML.reports.encoding_reports.RelevantSequenceExporter module¶
- class immuneML.reports.encoding_reports.RelevantSequenceExporter.RelevantSequenceExporter(dataset: RepertoireDataset = None, result_path: Path = None, name: str = None, number_of_processes: int = 1)[source]¶
Bases:
EncodingReportExports the sequences that are extracted as label-associated when using the
SequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoderin AIRR-compliant format.YAML specification:
definitions: reports: my_relevant_sequences: RelevantSequenceExporter
- classmethod build_object(**kwargs)[source]¶
Creates the object of the subclass of the Report class from the parameters so that it can be used in the analysis. Depending on the type of the report, the parameters provided here will be provided in parsing time, while the other necessary parameters (e.g., subset of the data from which the report should be created) will be provided at runtime. For more details, see specific direct subclasses of this class, describing different types of reports.
- Parameters:
**kwargs – keyword arguments that will be provided by users in the specification (if immuneML is used as a command line tool) or in the dictionary when calling the method from the code, and which should be used to create the report object
- Returns:
the object of the appropriate report class
- check_prerequisites()[source]¶
Checks prerequisites for the generation of the report of specific class (e.g., if the class of the MLMethod instance is the one required by the report, if the data has been encoded to make a report of encoded dataset). In the instructions in immuneML, this function is used to determine whether to call generate_report() in the specific situation. Each report subclass has its own set of prerequisites. If the report cannot be run, the information on this will be logged and the report skipped in the specific situation. No error will be raised. See subclasses of the class
Instructionfor more information on how the reports are executed.- Returns:
boolean value True if the prerequisites are o.k., and False otherwise.