Manuscript use case 1: Reproduction of a published study inside immuneML¶
In this use case, we show how the study by Emerson and colleagues on CMV status prediction from TCRbeta repertoires (Emerson et al. 2017) can be reproduced within immuneML. Additionally, we test the approach on datasets subsampled from the original study (to include randomly chosen 400, 200, 100 and 50 subjects) and estimate the performance of the approach when fewer examples are available.
The dataset was downloaded from Adaptive Biotechnologies’ website. Out of the 786 subjects (cohort 1: 666, cohort 2: 120), we removed 103 subjects from cohort 1 (1 with missing repertoire data, 25 with unknown CMV status, 3 with negative template counts for some of the sequences and the rest with no template count information).
The complete collection of original files used in this use case can be found in the NIRD research data archive (DOI: 10.11582/2021.00008). This also includes the metadata files for cohorts 1 and 2 with the list of subjects included in this use case. Note that the YAML specifications in the original dataset were compatible with immuneML version 1.0.1. This documentation page contains the YAML specifications for equivalent analyses with the latest immuneML version.
Reproduction of the CMV status predictions study¶
To reproduce the analysis, we used immuneML as a command line tool. We define the dataset to be used, data representation (encoding) which we call SequenceAbundance, the statistical model called ProbabilisticBinaryClassifier and different reports.
The encoding represents each repertoire by two numbers: the number of disease-associated sequences as determined by the Fisher’s exact test and the total number of sequences in the repertoire. As a hyperparameter of the encoding, it is possible to set the p-value threshold which can be used to determine which sequences are disease-associated. As in the original publication, we try out different p-value thresholds through cross-validation.
The statistical model ProbabilisticBinaryClassifier relies on SequenceAbundance encoding and fits two sets of parameters ({\(\alpha_0\), \(\beta_0\)} and {\(\alpha_1\), \(\beta_1\)}) to describe beta-distributed prior for CMV-negative and CMV-positive subjects. These parameters are then used to create log-posterior odds ratio for class assignment for new subjects.
Note
When used on large datasets, ‘SequenceAbundance’ encoder might be slow. If you want to reproduce the analysis faster and you are using immuneML version 2.2.0 or later, use CompAIRRSequenceAbundance encoder instead. p_value_threshold parameter is the same, and by default the analysis is performed using the amino acid sequence, and V and J genes. This can be turned of by setting ignore_genes to True (it is False by default).
To find the optimal p-value threshold we used 10-fold cross-validation on the cohort 1 and chose the one minimizing the cross-entropy loss (also called logarithmic loss). We then tested the performance of the optimal model (optimal p-value and the classifier fitted on resulting data representation) on the cohort 2 (as it was done in the original study).
The full YAML specification:
emerson_reproduction.yaml
definitions: datasets: cmv2017: # the full dataset including cohort 1 and cohort 2 format: ImmunoSEQRearrangement params: path: ../emerson_adaptive/ # dataset files as provided by Adaptive Biotechnologies unzipped into emerson_adaptive folder metadata_file: cmv_metadata.csv # metadata file including subjects for cohorts 1 and 2 result_path: imported_data/ # where to store imported data [immuneML implementation detail] encodings: # multiple encodings are defined that differ only on the p-value threshold they use to determine CMV-associated sequences enc01: SequenceAbundance: comparison_attributes: # how we define a sequence: as a combination of CDR3, V and J gene (as in the original publication) - sequence_aas - v_genes - j_genes p_value_threshold: 0.01 sequence_batch_size: 1000000 # immuneML implementation details repertoire_batch_size: 150 # immuneML implementation details enc001: # second encoding with different p-value SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.001 sequence_batch_size: 1000000 repertoire_batch_size: 150 enc0001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.0001 sequence_batch_size: 1000000 repertoire_batch_size: 150 enc00001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.00001 sequence_batch_size: 1000000 repertoire_batch_size: 150 enc000001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.000001 sequence_batch_size: 1000000 repertoire_batch_size: 150 ml_methods: # here we define the classifier ml: ProbabilisticBinaryClassifier: max_iterations: 50000 # how many iterations are allowed while estimating the distribution parameters update_rate: 0.01 reports: enc_data: DesignMatrixExporter # export encoded data in csv format sequence_association_likelihood: SequenceAssociationLikelihood # plot sequence association likelihood with estimated parameters feature_performance_plot: # show how performance changes depending on p-value threshold CVFeaturePerformance: feature: p_value_threshold sequence_overlap: # show if CMV-associated sequences overlap between CV folds DiseaseAssociatedSequenceCVOverlap: compare_in_selection: True compare_in_assessment: True relevant_sequences: RelevantSequenceExporter # export CMV-associated sequences emerson_reference_overlap: # check how many sequences overlap with the original study ReferenceSequenceOverlap: reference_path: emerson_reference.csv comparison_attributes: - sequence_aas - v_genes - j_genes instructions: cmv_study_reproduction: # defines what analysis should be like reports: [feature_performance_plot, sequence_overlap, emerson_reference_overlap] # reports to run after nested CV is finished assessment: # outer loop of nested cross-validation: split manually to training (cohort 1) and test (cohort 2) split_strategy: manual split_count: 1 manual_config: train_metadata_path: cmv_train_metadata.csv # cohort 1 test_metadata_path: cmv_test_metadata.csv # cohort 2 reports: encoding: [enc_data, relevant_sequences] models: [sequence_association_likelihood] selection: # inner loop of nested cross-validation performing 10 cross-validation to choose best p-value threshold split_strategy: k_fold split_count: 10 reports: encoding: [enc_data, relevant_sequences] number_of_processes: 32 dataset: cmv2017 labels: # which labels to use from the metadata, here: CMV - CMV: positive_class: True # positive class is called "True" - implementation detail used for encodings and some reports optimization_metric: log_loss # which metric to use for choose the best encoding metrics: # additional metrics to compute - balanced_accuracy - auc strategy: GridSearch # try out all settings combinations (all encodings here) type: TrainMLModel # name of the instruction refit_optimal_model: True # whether to refit the data on the whole dataset when all training and testing is finished settings: # combinations of encodings and classifiers to try out, basically, just listing all encodings with different p-values - encoding: enc01 ml_method: ml - encoding: enc001 ml_method: ml - encoding: enc0001 ml_method: ml - encoding: enc00001 ml_method: ml - encoding: enc000001 ml_method: ml output: format: HTML # output the result as HTML
Robustness assessment¶
After reproducing the study, we also assessed the robustness of the method on smaller datasets. To do that, we first constructed smaller datasets, and and then reproduced the analysis on those smaller datasets.
Constructing subsampled datasets¶
To construct smaller datasets of 400, 200, 100 and 50 subjects randomly from both cohorts, we used Subsampling instruction with the following YAML specification.
subsample_datasets.yaml
definitions: datasets: cmv2017: # we import the full dataset with 683 subjects as it was imported previously in immuneML-optimized format format: ImmuneML params: path: imported_data/cmv2017.yaml # Note that in older versions of immuneML (<3), immuneML datasets had .iml_dataset extension instead of .yaml instructions: subsampling_inst: # user-defined name of the instruction type: Subsampling # which instruction to execute dataset: cmv2017 # original dataset to be subsampled subsampled_dataset_sizes: # how large the subsampled datasets should be, one dataset will be created for each list item - 400 - 200 - 100 - 50 dataset_export_formats: # in which formats to export the subsampled datasets - ImmuneML
Running the analysis on subsampled datasets¶
To analyze and compare performances on datasets of different sizes, we use MultiDatasetBenchmarkTool.
The MultiDatasetBenchmarkTool can be run from the command line by providing the tool parameter in addition to YAML specification and the resulting folder:
immune-ml robustness_assessment_specs.yaml robustness_assessment_result/ --tool MultiDatasetBenchmarkTool
The YAML specification is mostly the same as when only TrainMLModel instruction is used except:
the dataset parameter is now called datasets and accepts a list of datasets on which the TrainMLModel instruction has to be performed (format and functionality are the same as described under reproduction), and
it has one additional parameter called benchmark_reports that will be executed after all datasets have been used to compare performances.
The YAML specification is given below.
robustness_assessment.yaml
definitions: datasets: # datasets for assessing robustness cmv2017_400: # with 400 repertoires format: ImmuneML params: path: subsampled_datasets/subsampling_inst/cmv2017_400_subsampled_1/exported/immuneml/cmv2017_400_subsampled_1.yaml # Note that in older versions of immuneML (<3), immuneML datasets had .iml_dataset extension instead of .yaml cmv2017_200: # with 200 repertoires format: ImmuneML params: path: subsampled_datasets/subsampling_inst/cmv2017_200_subsampled_2/exported/immuneml/cmv2017_200_subsampled_2.yaml cmv2017_100: # with 100 repertoires format: ImmuneML params: path: subsampled_datasets/subsampling_inst/cmv2017_100_subsampled_3/exported/immuneml/cmv2017_100_subsampled_3.yaml cmv2017_50: # with 50 repertoires format: ImmuneML params: path: subsampled_datasets/subsampling_inst/cmv2017_50_subsampled_4/exported/immuneml/cmv2017_50_subsampled_4.yaml encodings: # encodings as in Emerson et al. 2017 with different p-values to discover disease-associated combination of amino acid sequence and V and J gene enc01: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.01 sequence_batch_size: 1000000 # implementation details not affecting the algorithm, only the speed repertoire_batch_size: 150 # implementation details not affecting the algorithm, only the speed enc001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.001 sequence_batch_size: 1000000 repertoire_batch_size: 150 enc0001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.0001 sequence_batch_size: 1000000 repertoire_batch_size: 150 enc00001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.00001 sequence_batch_size: 1000000 repertoire_batch_size: 150 enc000001: SequenceAbundance: comparison_attributes: - sequence_aas - v_genes - j_genes p_value_threshold: 0.000001 sequence_batch_size: 1000000 repertoire_batch_size: 150 ml_methods: ml: ProbabilisticBinaryClassifier: # classifier as described in Emerson et al. 2017 max_iterations: 50000 update_rate: 0.01 reports: enc_data: DesignMatrixExporter # export encoded data as a csv file, also export labels and other info sequence_association_likelihood: SequenceAssociationLikelihood feature_performance_plot: # show how performance changes on average for different p-value thresholds for determining disease-associated sequences CVFeaturePerformance: feature: p_value_threshold is_feature_axis_categorical: True sequence_overlap: # check how stable are the estimates of disease-associated sequences across folds DiseaseAssociatedSequenceCVOverlap: compare_in_selection: True # compare disease-associated sequences only for the chosen optimal model across CV folds compare_in_assessment: True relevant_sequences: RelevantSequenceExporter # export disease-associated sequences emerson_reference_overlap: # check how much discovered disease-associated sequences overlap with the results published in the paper ReferenceSequenceOverlap: reference_path: emerson_reference.csv comparison_attributes: - sequence_aas - v_genes - j_genes sequence_overlap_across_datasets: DiseaseAssociatedSequenceOverlap # check how much disease-associated sequences overlap across datasets of different size performance_report: PerformanceOverview # show AUROC, AUPRC across datasets instructions: cmv_study_reproduction: # the format of the instruction is the same as above except there is a parameter benchmark_reports which are run when the instructions have finished reports: [feature_performance_plot, sequence_overlap, emerson_reference_overlap] # reports to run after nested CV is finished benchmark_reports: [sequence_overlap_across_datasets, performance_report] # reports to run after all dataset have been benchmarked assessment: # nested 5-fold CV (outer loop) split_strategy: k_fold split_count: 5 reports: encoding: [enc_data, relevant_sequences] models: [sequence_association_likelihood] selection: # nested 5-fold CV (inner loop) split_strategy: k_fold split_count: 5 reports: encoding: [enc_data, relevant_sequences] number_of_processes: 32 datasets: # instead of one dataset, there are 4 now when we use MultiDatasetBenchmarkTool - cmv2017_400 - cmv2017_200 - cmv2017_100 - cmv2017_50 labels: - CMV: positive_class: True metrics: - balanced_accuracy - auc optimization_metric: log_loss strategy: GridSearch # try out all combinations of encoding and ml_method listed under settings type: TrainMLModel # the type of the instruction which will be executed for each dataset refit_optimal_model: False settings: # combinations to try out to choose the best p-value - encoding: enc01 ml_method: ml - encoding: enc001 ml_method: ml - encoding: enc0001 ml_method: ml - encoding: enc00001 ml_method: ml output: format: HTML
Results¶
The results of reproducing the study by Emerson et al. are shown in the following figure:
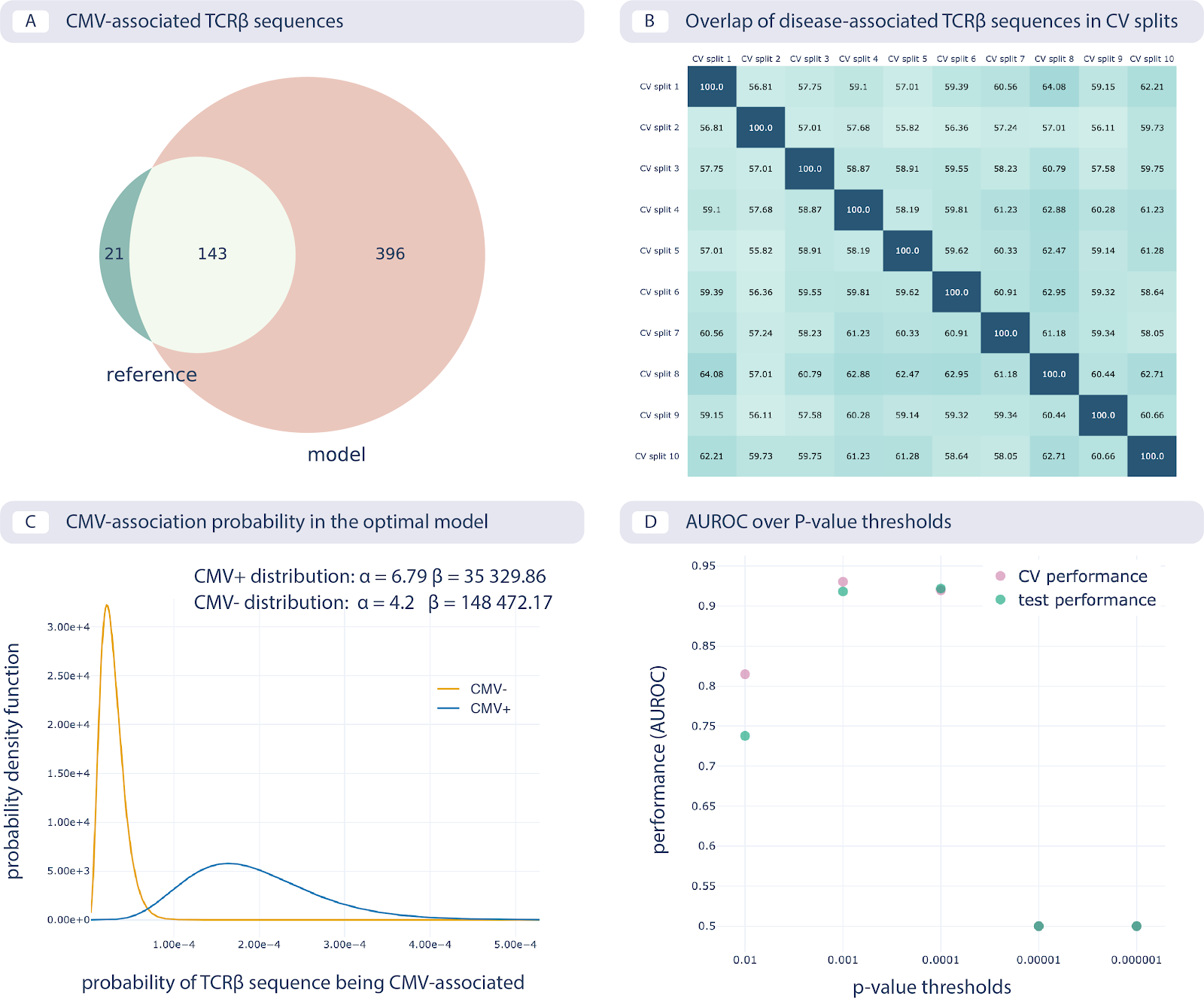
Reproducing the CMV status prediction study by Emerson et al.5 A. The overlap of the 164 disease-associated TCRβ sequences (V-TCRβaa-J) determined in the original study by Emerson et al., labeled “reference”, with those determined by the optimal model as reproduced here with a p-value threshold of 0.001 (labeled “model”). B. The overlap percentage of disease-associated TCRβ sequences for the optimal model with the p-value threshold of 0.001 between different data splits in 10-fold cross-validation (between 50% and 65% overlap). C. The probability that a TCRβ sequence is CMV-associated following a beta distribution estimated separately for CMV positive and negative subjects, which is then used for CMV status prediction of new subjects. D. Area under the ROC curve (AUROC) over p-value thresholds in training data (average AUROC over 10 cross-validation splits) and test data (AUROC in cohort 2).¶
Furthermore, the results of the robustness assessment (reproducing the study with a lower number of repertoires) are show here:
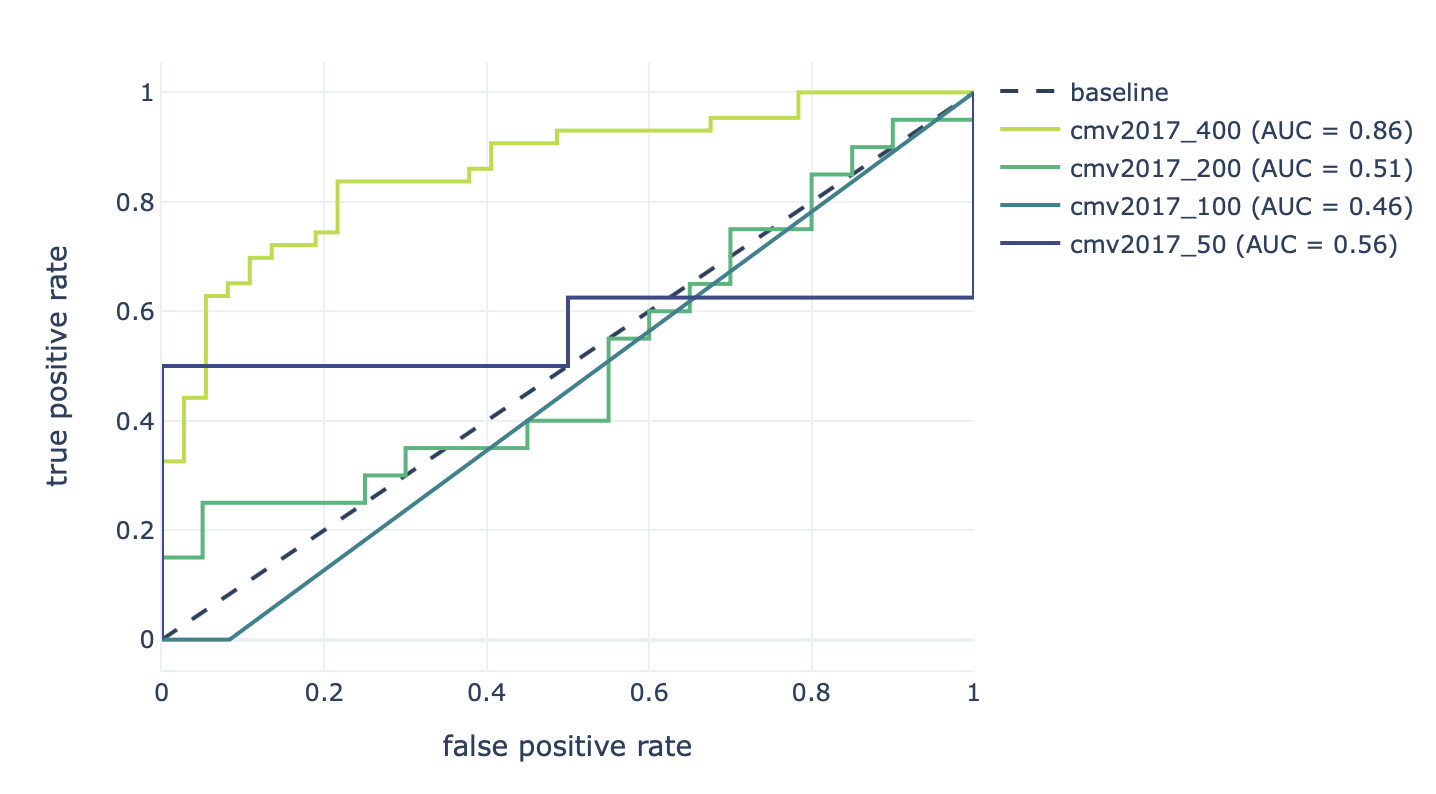
Decreasing the number of repertoires (400, 200, 100, and 50) leads to decreased prediction accuracy (AUROC: 0.86–0.46).¶