Report parameters¶
Under the definitions/reports component, the user can specify reports which visualise or summarise different properties
of the dataset or analysis. To use a report, it must be specified in the specific reports list of the instruction that generates it.
Reports have been divided into different types. Different types of reports can be specified depending on which instruction is run. Click on the name of the report type to see more details.
Data reports show some type of features or statistics about a given dataset.
Encoding reports show some type of features or statistics about an encoded dataset, or may export relevant sequences or tables.
ML model reports show some type of features or statistics about a single trained ML model (e.g., model coefficients).
Train ML model reports plot general statistics or export data of multiple models simultaneously when running the TrainMLModel instruction (e.g., performance comparison between models).
Multi dataset reports are special reports that can be specified when running immuneML with the
MultiDatasetBenchmarkTool. See Manuscript use case 1: Robustness assessment for an example.
Data reports¶
Data reports show some type of features or statistics about a given dataset.
When running the TrainMLModel instruction, data reports can be specified inside the ‘selection’ or ‘assessment’ specification under the keys ‘reports/data’ (current cross-validation split) or ‘reports/data_splits’ (train/test sub-splits). Example:
definitions:
reports:
my_data_report: SequenceCountDistribution
my_instruction:
type: TrainMLModel
selection:
reports:
data:
- my_data_report
# other parameters...
assessment:
reports:
data:
- my_data_report
# other parameters...
# other parameters...
Alternatively, when running the ExploratoryAnalysis instruction, data reports can be specified under ‘report’. Example:
my_instruction:
type: ExploratoryAnalysis
analyses:
my_first_analysis:
report: my_data_report
# other parameters...
# other parameters...
AminoAcidFrequencyDistribution¶
Generates a barplot showing the relative frequency of each amino acid at each position in the sequences of a dataset.
Example output:
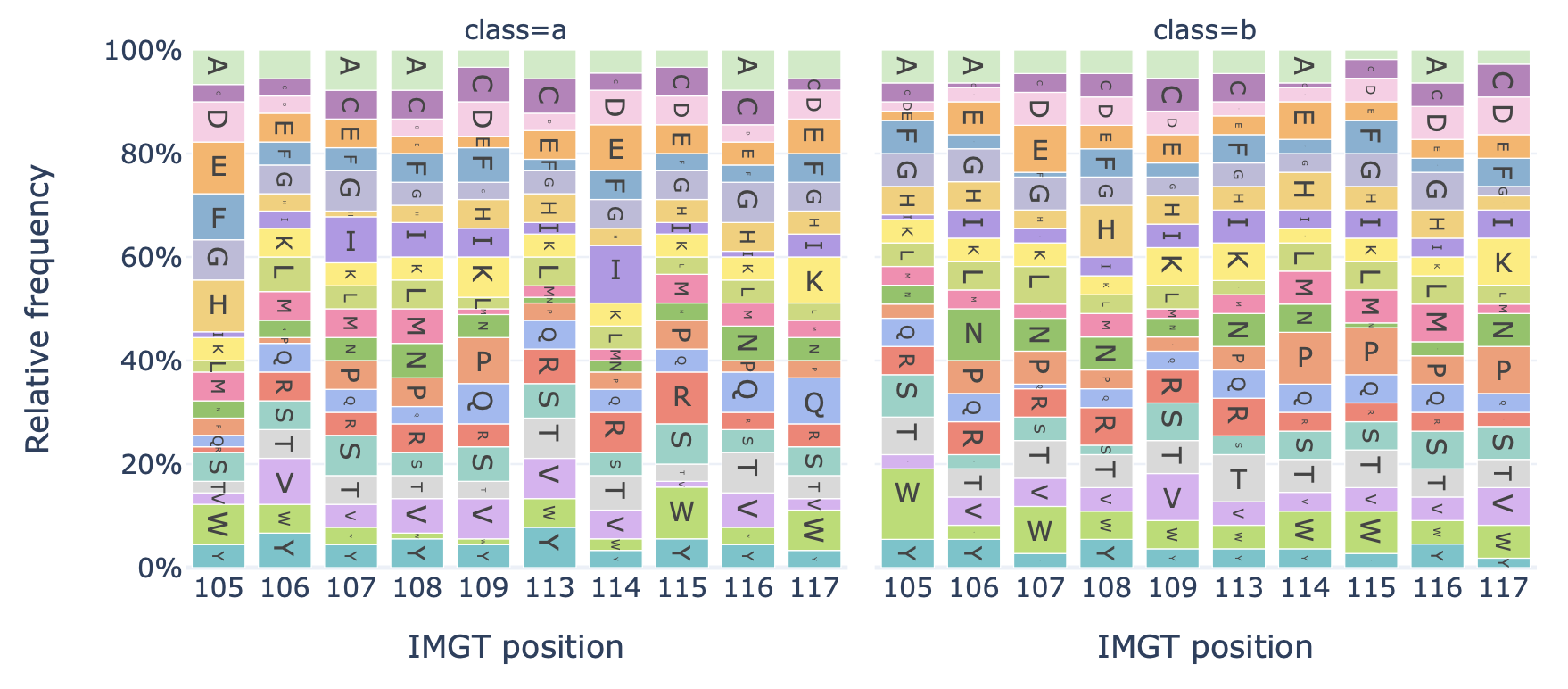
Specification arguments:
alignment (str): Alignment style for aligning sequences of different lengths. Options are as follows:
CENTER: center-align sequences of different lengths. The middle amino acid of any sequence be labelled position 0. By default, alignment is CENTER.
LEFT: left-align sequences of different lengths, starting at 0.
RIGHT: right align sequences of different lengths, ending at 0 (counting towards negative numbers).
IMGT: align sequences based on their IMGT positional numbering, considering the sequence region_type (IMGT_CDR3 or IMGT_JUNCTION). The main difference between CENTER and IMGT is that IMGT aligns the first and last amino acids, adding gaps in the middle, whereas CENTER aligns the middle of the sequences, padding with gaps at the start and end of the sequence. When region_type is IMGT_JUNCTION, the IMGT positions run from 104 (conserved C) to 118 (conserved W/F). When IMGT_CDR3 is used, these positions are 105 to 117. For long CDR3 sequences, additional numbers are added in between IMGT positions 111 and 112. See the official IMGT documentation for more details: https://www.imgt.org/IMGTScientificChart/Numbering/CDR3-IMGTgaps.html
relative_frequency (bool): Whether to plot relative frequencies (true) or absolute counts (false) of the positional amino acids. Note that when sequences are of different length, setting relative_frequency to True will produce different results depending on the alignment type, as some positions are only covered by the longest sequences. By default, relative_frequency is False.
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. If split_by_label is set to true, the percentage-wise frequency difference between classes is plotted additionally. By default, split_by_label is False.
label (str): if split_by_label is set to True, a label can be specified here.
region_type (str): which part of the sequence to check; e.g., IMGT_CDR3
YAML specification:
definitions:
reports:
my_aa_freq_report:
AminoAcidFrequencyDistribution:
relative_frequency: False
split_by_label: True
label: CMV
region_type: IMGT_CDR3
CompAIRRClusteringReport¶
A report that uses CompAIRR to compute repertoire distances based on sequence overlap and performs hierarchical clustering on the resulting distance matrix. The clustering results are visualized using a dendrogram, colored by a specified label.
The report assumes that CompAIRR (https://github.com/uio-bmi/compairr) has been installed.
Note
This is an experimental feature.
Specification arguments:
labels (list): The list of labels to highlight below the dendrogram. The labels should be present in the dataset.
compairr_path (str): Path to the CompAIRR executable.
indels (bool): Whether to allow insertions/deletions when matching sequences (default: False)
ignore_counts (bool): Whether to ignore sequence counts when computing overlap (default: False)
ignore_genes (bool): Whether to ignore V/J gene assignments when matching sequences (default: False)
threads (int): Number of threads to use for CompAIRR computation (default: 4)
linkage_method (str): The linkage method to use for hierarchical clustering (default: ‘single’)
is_cdr3 (bool): Whether the sequences represent CDR3s (default: True)
clustering_criterion (str): The criterion to use for clustering (default: ‘distance’), as defined in scipy.cluster.hierarchy.linkage; valid values are ‘distance’, ‘maxclust’, ‘monocrit’, ‘maxclust_monocrit’
clustering_threshold (float): The threshold for the clustering algorithm (default: 0.5), mapped to ‘t’ parameter in scipy.cluster.hierarchy.fcluster
YAML specification:
definitions:
reports:
my_compairr_clustering_report:
CompAIRRClusteringReport:
labels: [disease]
compairr_path: /path/to/compairr
indels: false
ignore_counts: true
ignore_genes: true
threads: 4
linkage_method: single
is_cdr3: true
clustering_criterion: distance
clustering_threshold: 0.5
GLIPH2Exporter¶
Report which exports the receptor data to GLIPH2 format so that it can be directly used in GLIPH2 tool. Currently, the report accepts only receptor datasets.
GLIPH2 publication: Huang H, Wang C, Rubelt F, Scriba TJ, Davis MM. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nature Biotechnology. Published online April 27, 2020:1-9. doi:10.1038/s41587-020-0505-4
Specification arguments:
condition (str): name of the parameter present in the receptor metadata in the dataset; condition can be anything which can be processed in GLIPH2, such as tissue type or treatment.
YAML specification:
definitions:
reports:
my_gliph2_exporter:
GLIPH2Exporter:
condition: epitope # for instance, epitope parameter is present in receptors' metadata with values such as "MtbLys" for Mycobacterium tuberculosis (as shown in the original paper).
LabelDist¶
LabelDist report plots the distribution of label values for all labels provided as input to the report.
Specification arguments:
labels (list): list of label names as they appear in the metadata file (RepertoireDataset) or in data files (Receptor/SequenceDataset).
YAML specification:
LabelOverlap¶
This report creates a heatmap where the columns are the values of one label and rows are the values of another label, and the cells contain the number of samples that have both label values. It works for any dataset type.
Specification arguments:
column_label (str): Name of the label to be used as columns in the heatmap.
row_label (str): Name of the label to be used as rows in the heatmap.
YAML specification:
my_data_report:
LabelOverlap:
column_label: epitope
row_label: batch
MotifGeneralizationAnalysis¶
This report splits the given dataset into a training and validation set, identifies significant motifs using the
MotifEncoder
on the training set and plots the precision/recall and precision/true positive predictions of motifs
on both the training and validation sets. This can be used to:
determine the optimal recall cutoff for motifs of a given size
investigate how well motifs learned on a training set generalize to a test set
After running this report and determining the optimal recall cutoffs, the report
MotifTestSetPerformance can be run to
plot the performance on an independent test set.
Note: the MotifEncoder (and thus this report) can only be used for sequences of the same length.
Specification arguments:
label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
training_set_identifier_path (str): Path to a file containing ‘sequence_identifiers’ of the sequences used for the training set. This file should have a single column named ‘example_id’ and have one sequence identifier per line. If training_set_identifier_path is not set, a random subset of the data (according to training_percentage) will be assigned to be the training set.
training_percentage (float): If training_set_identifier_path is not set, this value is used to specify the fraction of sequences that will be randomly assigned to form the training set. Should be a value between 0 and 1. By default, training_percentage is 0.7.
random_seed (int): Random seed for splitting the data into training and validation sets a training_set_identifier_path is not provided.
split_by_motif_size (bool): Whether to split the analysis per motif size. If true, a recall threshold is learned for each motif size, and figures are generated for each motif size independently. By default, split_by_motif_size is true.
min_precision:
MotifEncoderparameter. The minimum precision threshold for keeping a motif on the training set. By default, min_precision is 0.9.test_precision_threshold (float). The desired precision on the test set, given that motifs are learned by using a training set with a precision threshold of min_precision. It is recommended for test_precision_threshold to be lower than min_precision, e.g., min_precision - 0.1. By default, test_precision_threshold is 0.8.
min_recall (float):
MotifEncoderparameter. The minimum recall threshold for keeping a motif. Any learned recall threshold will be at least as high as the set min_recall value. The default value for min_recall is 0.min_true_positives (int):
MotifEncoderparameter. The minimum number of true positive training sequences that a motif needs to occur in. The default value for min_true_positives is 1.max_positions (int):
MotifEncoderparameter. The maximum motif size. This is number of positional amino acids the motif consists of (excluding gaps). The default value for max_positions is 4.min_positions (int):
MotifEncoderparameter. The minimum motif size (see also: max_positions). The default value for min_positions is 1.no_gaps (bool):
MotifEncoderparameter. Must be set to True if only contiguous motifs (position-specific k-mers) are allowed. By default, no_gaps is False, meaning both gapped and ungapped motifs are searched for.smoothen_combined_precision (bool): whether to add a smoothed line representing the combined precision to the precision-vs-TP plot. When set to True, this may take considerable extra time to compute. By default, plot_smoothed_combined_precision is set to True.
min_points_in_window (int): Parameter for smoothing the combined_precision line in the precision-vs-TP plot through lognormal kernel smoothing with adaptive window size. This parameter determines the minimum number of points that need to be present in a window to determine the adaptive window size. By default, min_points_in_window is 50.
smoothing_constant1: Parameter for smoothing the combined_precision line in the precision-vs-TP plot through lognormal kernel smoothing with adaptive window size. This smoothing constant determines the dependence of the smoothness on the window size. Increasing this increases smoothness for regions where few points are present. By default, smoothing_constant1 is 5.
smoothing_constant2: Parameter for smoothing the combined_precision line in the precision-vs-TP plot through lognormal kernel smoothing. with adaptive window size. This smoothing constant can be used to scale the overall kernel width, thus influencing the smoothness of all regions regardless of data density. By default, smoothing_constant2 is 10.
training_set_name (str): Name of the training set to be used in figures. By default, the training_set_name is ‘training set’.
test_set_name (str): Name of the test set to be used in figures. By default, the test_set_name is ‘test set’.
highlight_motifs_path (str): Path to a set of motifs of interest to highlight in the output figures (such as implanted ground-truth motifs). By default, no motifs are highlighted.
highlight_motifs_name (str): IF highlight_motifs_path is defined, this name will be used to label the motifs of interest in the output figures.
YAML specification:
definitions:
reports:
my_motif_generalization:
MotifGeneralizationAnalysis:
min_precision: 0.9
min_recall: 0.1
label: # Define a label, and the positive class for that given label
CMV:
positive_class: +
NodeDegreeDistribution¶
A report that uses CompAIRR to compute the node degree distribution of a sequence dataset. Results are visualized as a histogram and stored in a TSV file.
The report assumes that CompAIRR (https://github.com/uio-bmi/compairr) has been installed.
Specification arguments:
compairr_path (str): The path to the CompAIRR executable.
region_type (str): The region type to analyze. Can be either “IMGT_CDR3” or “IMGT_JUNCTION”.
indels (bool): Whether to include indels in the analysis. Default is False.
ignore_genes (bool): Whether to ignore gene names in the analysis. Default is False.
hamming_distance (int): The Hamming distance to use for the analysis. Default is 1.
per_repertoire (bool): Whether to compute the node degree distribution for each repertoire separately. Only applicable when using a RepertoireDataset. Default is False.
per_label (bool): Whether to compute the node degree distribution for each label separately. Only applicable when using a RepertoireDataset. Default is False.
threads (int): The number of threads to use for the analysis. Default is 4.
YAML specification:
NodeDegreeDistribution:
compairr_path: /path/to/compairr
region_type: IMGT_JUNCTION
indels: False
ignore_genes: False
hamming_distance: 1
per_repertoire: False
per_label: False
threads: 4
RecoveredSignificantFeatures¶
Compares a given collection of ground truth implanted signals (sequences or k-mers) to the significant label-associated k-mers or sequences according to Fisher’s exact test.
Internally uses the KmerAbundanceEncoder for calculating
significant k-mers, and
SequenceAbundanceEncoder or
CompAIRRSequenceAbundanceEncoder
to calculate significant full sequences (depending on whether the argument compairr_path was set).
This report creates two plots:
the first plot is a bar chart showing what percentage of the ground truth implanted signals were found to be significant.
the second plot is a bar chart showing what percentage of the k-mers/sequences found to be significant match the ground truth implanted signals.
To compare k-mers or sequences of differing lengths, the ground truth sequences or long k-mers are split into k-mers of the given size through a sliding window approach. When comparing ‘full_sequences’ to ground truth sequences, a match is only registered if both sequences are of equal length.
Specification arguments:
ground_truth_sequences_path (str): Path to a file containing the true implanted (sub)sequences, e.g., full sequences or k-mers. The file should contain one sequence per line, without a header, and without V or J genes.
sequence_type (str): either amino acid or nucleotide; which type of sequence to use for the analysis
region_type (str): which AIRR field to use for comparison, e.g. IMGT_CDR3 or IMGT_JUNCTION
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. When using a full sequence encoding (SequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoder), specify ‘full_sequence’ here. Each value specified under k_values will represent one bar in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
compairr_path (str): If ‘full_sequence’ is listed under k_values, the path to the CompAIRR executable may be provided. If the compairr_path is specified, the
CompAIRRSequenceAbundanceEncoderwill be used to compute the significant sequences. If the path is not specified and ‘full_sequence’ is listed under k-values,SequenceAbundanceEncoderwill be used.
YAML specification:
definitions:
reports:
my_recovered_significant_features_report:
RecoveredSignificantFeatures:
groundtruth_sequences_path: path/to/groundtruth/sequences.txt
trim_leading_trailing: False
p_values:
- 0.1
- 0.01
- 0.001
- 0.0001
k_values:
- 3
- 4
- 5
- full_sequence
compairr_path: path/to/compairr # can be specified if 'full_sequence' is listed under k_values
label: # Define a label, and the positive class for that given label
CMV:
positive_class: +
RepertoireClonotypeSummary¶
Shows the number of distinct clonotypes per repertoire in a given dataset as a bar plot.
Specification arguments:
color_label (str): the label to color the bar plot by (optional, default: None)
facet_label (str): the label to facet the bar plot by (optional, default: None)
YAML specification:
definitions:
reports:
my_clonotype_summary_rep:
RepertoireClonotypeSummary:
color_label: celiac
facet_label: hla
SequenceCountDistribution¶
Generates a histogram of the duplicate counts of the sequences in a dataset.
Specification arguments:
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. By default, split_by_label is False.
label (str): Optional label for separating the results by color/creating separate plots. Note that this should the name of a valid dataset label.
YAML specification:
my_sld_report:
SequenceCountDistribution:
label: disease
SequenceLengthDistribution¶
Generates a histogram of the lengths of the sequences in a dataset.
Specification arguments:
sequence_type (str): whether to check the length of amino acid or nucleotide sequences; default value is ‘amino_acid’
region_type (str): which part of the sequence to examine; e.g., IMGT_CDR3
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. By default, split_by_label is False.
label (str): if split_by_label is set to True, a label can be specified here.
plot_frequencies (bool): if set to True, the plot will show the frequencies of the sequence lengths instead of the counts. By default, plot_frequencies is False.
YAML specification:
definitions:
reports:
my_sld_report:
SequenceLengthDistribution:
sequence_type: amino_acid
region_type: IMGT_CDR3
label: label_1
split_by_label: True
plot_frequencies: True
SequencesWithSignificantKmers¶
Given a list of reference sequences, this report writes out the subsets of reference sequences containing significant k-mers
(as computed by the KmerAbundanceEncoder using Fisher’s exact test).
For each combination of p-value and k-mer size given, a file is written containing all sequences containing a significant k-mer of the given size at the given p-value.
Specification arguments:
reference_sequences_path (str): Path to a file containing the reference sequences, The file should contain one sequence per line, without a header, and without V or J genes.
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. Each k-mer length will become one panel in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
YAML specification:
definitions:
reports:
my_sequences_with_significant_kmers:
SequencesWithSignificantKmers:
reference_sequences_path: path/to/reference/sequences.txt
p_values:
- 0.1
- 0.01
- 0.001
- 0.0001
k_values:
- 3
- 4
- 5
label: # Define a label, and the positive class for that given label
CMV:
positive_class: +
ShannonDiversityOverview¶
Computes Shannon diversity for each repertoire using Shannon diversity encoder and plots the results in a histogram, optionally stratified by labels.
Dataset type:
Repertoire Dataset
Specification arguments:
color_label (str): The label used to color the histogram bars. Default is None.
facet_row_label (str): The label used to facet the histogram into multiple rows. Default is None, meaning no row faceting.
facet_col_label (str): The label used to facet the histogram into multiple columns. Default is None, meaning no column faceting.
YAML specification:
definitions:
reports:
shannon_div_rep:
ShannonDiversityOverview:
color_label: disease_status
SignificantFeatures¶
Plots a boxplot of the number of significant features (label-associated k-mers or sequences) per Repertoire according to Fisher’s exact test, across different classes for the given label.
Internally uses the KmerAbundanceEncoder for calculating
significant k-mers, and
SequenceAbundanceEncoder or
CompAIRRSequenceAbundanceEncoder
to calculate significant full sequences (depending on whether the argument compairr_path was set).
Specification arguments:
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. When using a full sequence encoding (SequenceAbundanceEncoderorCompAIRRSequenceAbundanceEncoder), specify ‘full_sequence’ here. Each value specified under k_values will represent one boxplot in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
compairr_path (str): If ‘full_sequence’ is listed under k_values, the path to the CompAIRR executable may be provided. If the compairr_path is specified, the
CompAIRRSequenceAbundanceEncoderwill be used to compute the significant sequences. If the path is not specified and ‘full_sequence’ is listed under k-values,SequenceAbundanceEncoderwill be used.log_scale (bool): Whether to plot the y axis in log10 scale (log_scale = True) or continuous scale (log_scale = False). By default, log_scale is False.
YAML specification:
definitions:
reports:
my_significant_features_report:
SignificantFeatures:
p_values:
- 0.1
- 0.01
- 0.001
- 0.0001
k_values:
- 3
- 4
- 5
- full_sequence
compairr_path: path/to/compairr # can be specified if 'full_sequence' is listed under k_values
label: # Define a label, and the positive class for that given label
CMV:
positive_class: +
log_scale: False
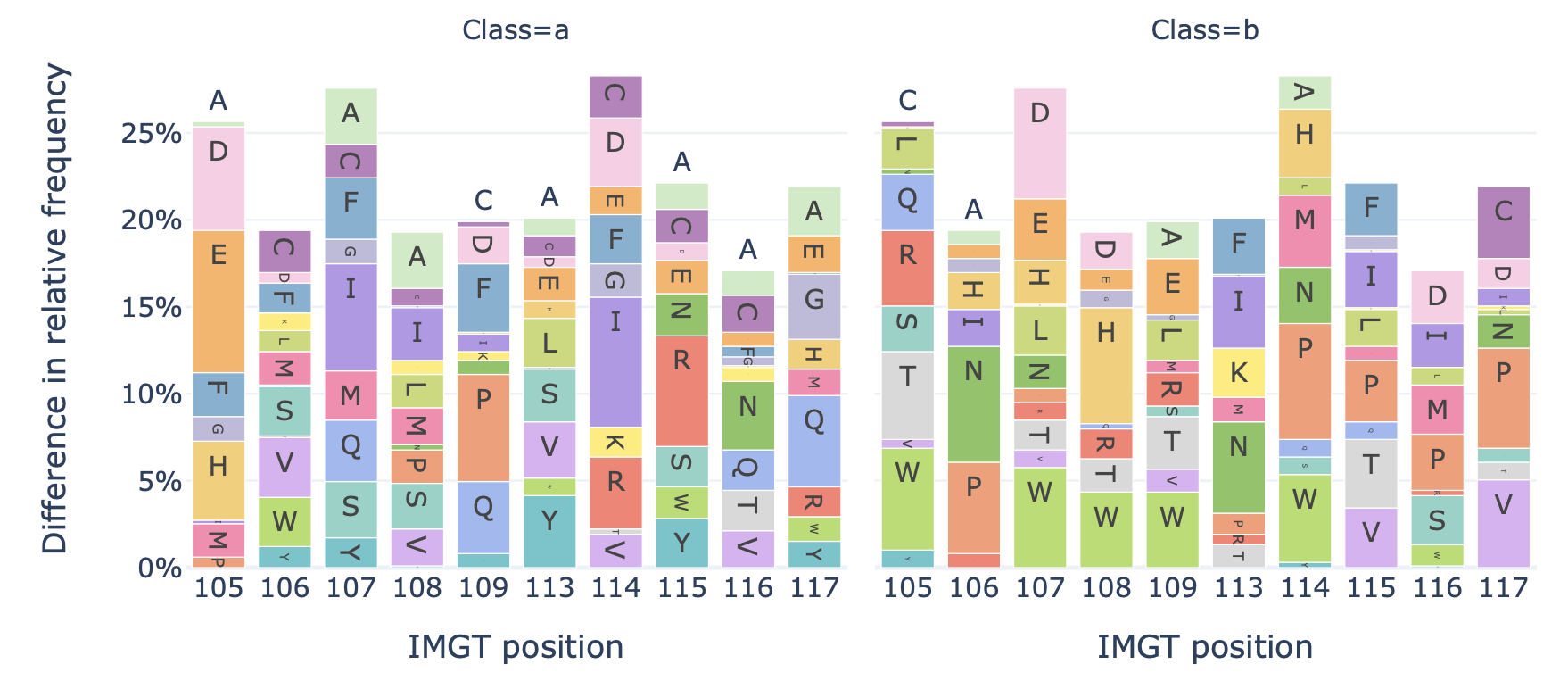
SignificantKmerPositions¶
Plots the number of significant k-mers (as computed by the KmerAbundanceEncoder using Fisher’s exact test)
observed at each IMGT position of a given list of reference sequences.
This report creates a stacked bar chart, where each bar represents an IMGT position, and each segment of the stack represents the observed frequency
of one ‘significant’ k-mer at that position.
Specification arguments:
reference_sequences_path (str): Path to a file containing the reference sequences, The file should contain one sequence per line, without a header, and without V or J genes.
p_values (list): The p value thresholds to be used by Fisher’s exact test. Each p-value specified here will become one panel in the output figure.
k_values (list): Length of the k-mers (number of amino acids) created by the
KmerAbundanceEncoder. Each k-mer length will become one panel in the output figure.label (dict): A label configuration. One label should be specified, and the positive_class for this label should be defined. See the YAML specification below for an example.
sequence_type (str): nucleotide or amino_acid
region_type (str): which AIRR field to consider, e.g., IMGT_CDR3 or IMGT_JUNCTION
YAML specification:
definitions:
reports:
my_significant_kmer_positions_report:
SignificantKmerPositions:
reference_sequences_path: path/to/reference/sequences.txt
p_values:
- 0.1
- 0.01
- 0.001
- 0.0001
k_values:
- 3
- 4
- 5
label: # Define a label, and the positive class for that given label
CMV:
positive_class: +
TrueMotifsSummaryBarplot¶
This report can be used to show how well motifs (for example, motifs introduced using the Simulation instruction) are learned across different generative models. The report shows a bar plot with the proportion of sequences in each dataset that contain the given motifs. Bars are grouped by the dataset origin (e.g., train, PWM, VAE, LSTM) and the signals provided. The report also shows how many of the sequences are memorized (seen in train data) and how many are novel (not seen in train data).
Specification arguments:
region_type (str): which part of the sequence to check; e.g., IMGT_CDR3
implanted_motifs_per_signal (dict): a nested dictionary that specifies the motif seeds that were implanted in the given dataset. The first level of keys in this dictionary represents the different signals. In the inner dictionary there should be two keys: “seeds” and “gap_sizes”.
seeds: a list of motif seeds. The seeds may contain gaps, specified by a ‘/’ symbol.
gap_sizes: a list of all the possible gap sizes that were used when implanting a gapped motif seed. When no gapped seeds are used, this value has no effect.
YAML specification:
definitions:
reports:
my_motif_report:
TrueMotifsSummaryBarplot:
region_type: IMGT_CDR3
implanted_motifs_per_signal:
my_signal1:
seeds:
- DEQ
gap_sizes:
- 0
my_signal2:
seeds:
- AS/G
gap_sizes:
- 2
VJGeneDistribution¶
This report creates several plots to gain insight into the V and J gene distribution of a given dataset. When a label is provided, the information in the plots is separated per label value, either by color or by creating separate plots. This way one can for example see if a particular V or J gene is more prevalent across disease associated receptors.
Individual V and J gene distributions: for sequence and receptor datasets, a bar plot is created showing how often each V or J gene occurs in the dataset. For repertoire datasets, boxplots are used to represent how often each V or J gene is used across all repertoires. Since repertoires may differ in size, these counts are normalised by the repertoire size (original count values are additionaly exported in tsv files).
Combined V and J gene distributions: for sequence and receptor datasets, a heatmap is created showing how often each combination of V and J genes occurs in the dataset. A similar plot is created for repertoire datasets, except in this case only the average value for the normalised gene usage frequencies are shown (original count values are additionaly exported in tsv files).
Specification arguments:
split_by_label (bool): Whether to split the plots by a label. If set to true, the Dataset must either contain a single label, or alternatively the label of interest can be specified under ‘label’. By default, split_by_label is False.
label (str): Optional label for separating the results by color/creating separate plots. Note that this should the name of a valid dataset label.
is_sequence_label (bool): for RepertoireDatasets, indicates if the label applies to the sequence level (e.g., antigen binding versus non-binding across repertoires) or repertoire level (e.g., diseased repertoires versus healthy repertoires). By default, is_sequence_label is False. For Sequence- and ReceptorDatasets, this parameter is ignored.
show_joint_dist (bool): whether to show the combined V and J gene distribution. Default is True.
YAML specification:
definitions:
reports:
my_vj_gene_report:
VJGeneDistribution:
label: ag_binding
show_joint_dist: false
Encoding reports¶
Encoding reports show some type of features or statistics about an encoded dataset, or may in some cases export relevant sequences or tables.
When running the TrainMLModel instruction, encoding reports can be specified inside the ‘selection’ or ‘assessment’ specification under the key ‘reports/encoding’. Example:
my_instruction:
type: TrainMLModel
selection:
reports:
encoding:
- my_encoding_report
# other parameters...
assessment:
reports:
encoding:
- my_encoding_report
# other parameters...
# other parameters...
Alternatively, when running the ExploratoryAnalysis instruction, encoding reports can be specified under ‘report’. Example:
my_instruction:
type: ExploratoryAnalysis
analyses:
my_first_analysis:
report: my_encoding_report
# other parameters...
# other parameters...
DesignMatrixExporter¶
Exports the design matrix and related information of a given encoded Dataset to csv files. If the encoded data has more than 2 dimensions (such as when using the OneHot encoder with option Flatten=False), the data are then exported to different formats to facilitate their import with external software.
Specification arguments:
file_format (str): the format and extension of the file to store the design matrix. The supported formats are: npy, csv, pt, hdf5, npy.zip, csv.zip or hdf5.zip.
Note: when using hdf5 or hdf5.zip output formats, make sure the ‘hdf5’ dependency is installed.
YAML specification:
definitions:
reports:
my_dme_report:
DesignMatrixExporter:
file_format: csv
DimensionalityReduction¶
This report visualizes the data obtained by dimensionality reduction. The data points can be highlighted by label of interest. It is also possible to specify labels that contain lists of values (e.g., HLA), in which case the data points will be duplicated (so that each point refers to one HLA allele) and jittered slightly to improve visibility before being highlighted by the concrete HLA allele values.
When a dim_red_method is configured, its components parameter determines which two components are plotted
and overrides the report-level components. When no dim_red_method is set (using pre-computed
dimensionality-reduced data), the report-level components selects which two columns to plot.
All computed components are always written to the output CSV.
For PCA the explained variance per component is exported to a separate CSV and annotated on the axis labels.
For KernelPCA with compute_total_variance: true the fraction of total kernel-space variance is shown instead.
Specification arguments:
labels (list): names of the label to use for highlighting data points; or None
components (list): which two components (1-indexed) to plot when no
dim_red_methodis provided. When adim_red_methodis set, use its owncomponentsparameter instead. Default: [1, 2].dim_red_method (str): dimensionality reduction method to be used for plotting; if set, in a workflow, this dimensionality reduction will be used for plotting instead of any other set in the workflow; if None, it will visualize the encoded data of reduced dimensionality if set
YAML specification:
definitions:
reports:
# Plot PC3 vs PC4 from a 5-component PCA, annotated with explained variance
rep1:
DimensionalityReduction:
labels: [epitope, source]
dim_red_method:
PCA:
n_components: 5
components: [3, 4]
# Plot components 1 vs 2 from pre-computed dimensionality-reduced data
rep2:
DimensionalityReduction:
labels: [epitope]
components: [1, 2]
FeatureComparison¶
Encoding a dataset results in a numeric matrix, where the rows are examples (e.g., sequences, receptors, repertoires) and the columns are features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
This report separates the examples based on a binary metadata label, and plots the mean feature value of each feature in one example group against the other example group (for example: plot the feature value of ‘sick’ repertoires on the x axis, and ‘healthy’ repertoires on the y axis to spot consistent differences). The plot can be separated into different colors or facets using other metadata labels (for example: plot the average feature values of ‘cohort1’, ‘cohort2’ and ‘cohort3’ in different colors to spot biases).
Alternatively, when plotting features without comparing them across a binary label, see:
FeatureValueBarplot report to plot
a simple bar chart per feature (average across examples).
Or FeatureDistribution report to plot
the distribution of each feature across examples, rather than only showing the mean value in a bar plot.
Example output:
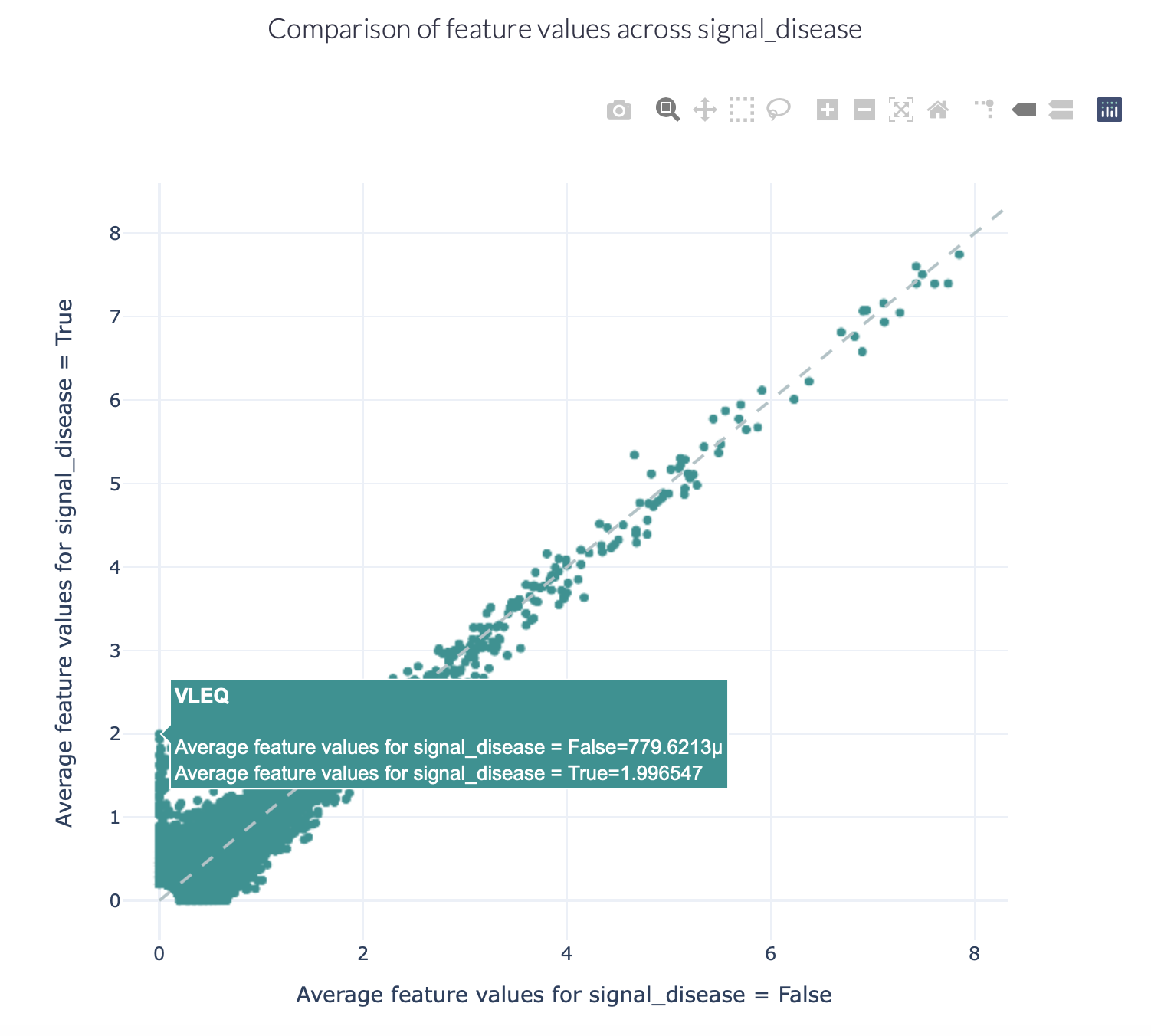
Specification arguments:
comparison_label (str): Mandatory label. This label is used to split the encoded data matrix and define the x and y axes of the plot. This label is only allowed to have 2 classes (for example: sick and healthy, binding and non-binding).
color_grouping_label (str): Optional label that is used to color the points in the scatterplot. This can not be the same as comparison_label.
row_grouping_label (str): Optional label that is used to group scatterplots into different row facets. This can not be the same as comparison_label.
column_grouping_label (str): Optional label that is used to group scatterplots into different column facets. This can not be the same as comparison_label.
show_error_bar (bool): Whether to show the error bar (standard deviation) for the points, both in the x and y dimension.
log_scale (bool): Whether to plot the x and y axes in log10 scale (log_scale = True) or continuous scale (log_scale = False). By default, log_scale is False.
keep_fraction (float): The total number of features may be very large and only the features differing significantly across comparison labels may be of interest. When the keep_fraction parameter is set below 1, only the fraction of features that differs the most across comparison labels is kept for plotting (note that the produced .csv file still contains all data). By default, keep_fraction is 1, meaning that all features are plotted.
opacity (float): a value between 0 and 1 setting the opacity for data points making it easier to see if there are overlapping points
error_function (str): which error function to use for the error bar. Options are ‘std’ (standard deviation) or ‘sem’ (standard error of the mean). Default: std.
YAML specification:
definitions:
reports:
my_comparison_report:
FeatureComparison: # compare the different classes defined in the label disease
comparison_label: disease
FeatureDistribution¶
Encoding a dataset results in a numeric matrix, where the rows are examples (e.g., sequences, receptors, repertoires) and the columns are features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
This report plots the distribution of feature values. For each feature, a violin plot is created to show the distribution of feature values across all examples. The violin plots can be separated into different colors or facets using metadata labels (for example: plot the feature distributions of ‘cohort1’, ‘cohort2’ and ‘cohort3’ in different colors to spot biases).
See also: FeatureValueBarplot report to plot
a simple bar chart per feature (average across examples), rather than the entire distribution.
Or FeatureComparison report to compare
features across binary metadata labels (e.g., plot the feature value of ‘sick’ repertoires on the x axis,
and ‘healthy’ repertoires on the y axis).
Example output:
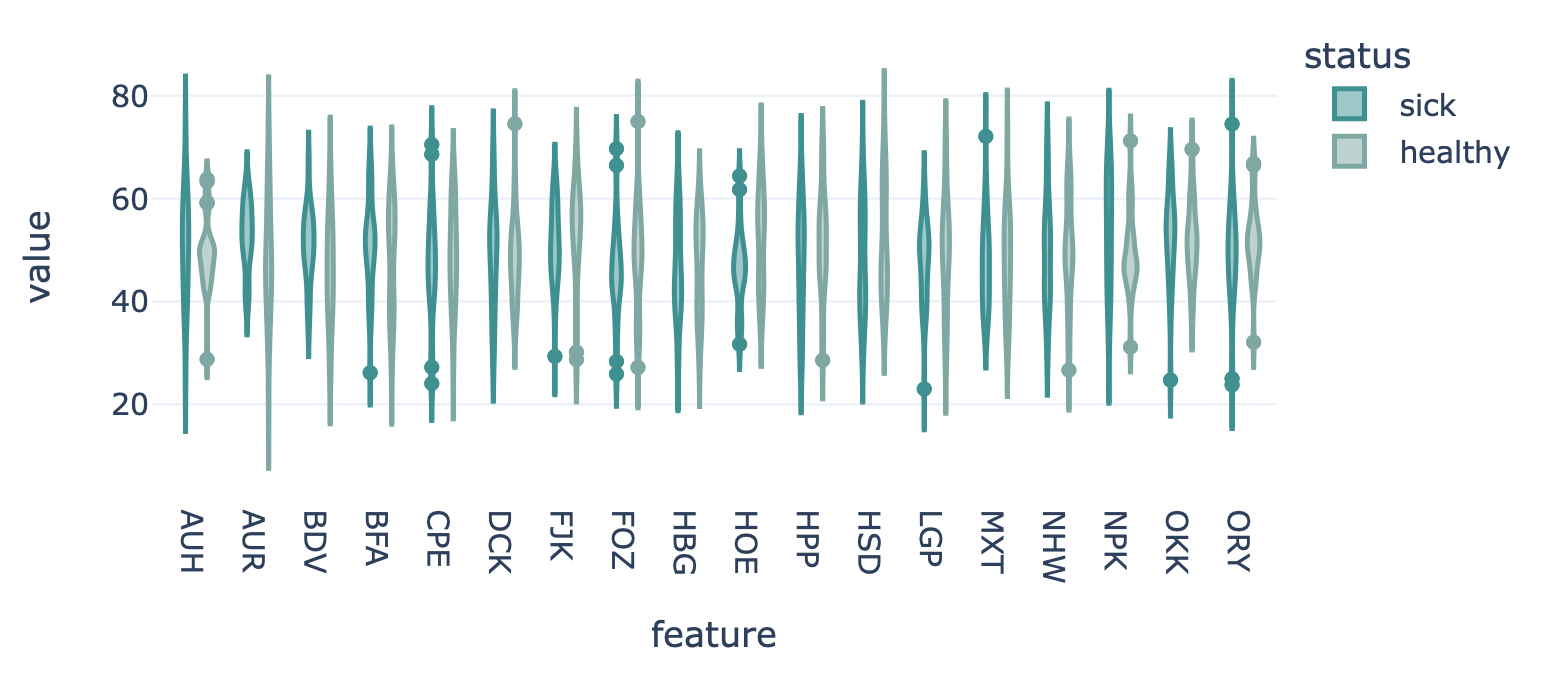
Specification arguments:
color_grouping_label (str): The label that is used to color each bar, at each level of the grouping_label.
row_grouping_label (str): The label that is used to group bars into different row facets.
column_grouping_label (str): The label that is used to group bars into different column facets.
mode (str): either ‘normal’, ‘sparse’ or ‘auto’ (default). in the ‘normal’ mode there are normal boxplots corresponding to each column of the encoded dataset matrix; in the ‘sparse’ mode all zero cells are eliminated before passing the data to the boxplots. If mode is set to ‘auto’, then it will automatically set to ‘sparse’ if the density of the matrix is below 0.01
x_title (str): x-axis label
y_title (str): y-axis label
plot_top_n (int): plot n of the largest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels.
plot_bottom_n (int): plot n of the smallest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels.
plot_all_features (bool): whether to plot all (might be slow for large number of features)
error_function (str): which error function to use for the error bar. Options are ‘std’ (standard deviation) or ‘sem’ (standard error of the mean). Default: std.
YAML specification:
definitions:
reports:
my_fdistr_report:
FeatureDistribution:
mode: sparse
plot_all_features: True
plot_top_n: 10
plot_bottom_n: 10
FeatureValueBarplot¶
Encoding a dataset results in a numeric matrix, where the rows are examples (e.g., sequences, receptors, repertoires) and the columns are features. For example, when KmerFrequency encoder is used, the features are the k-mers (AAA, AAC, etc..) and the feature values are the frequencies per k-mer.
This report plots the mean feature values per feature. A bar plot is created where the average feature value across all examples is shown, with optional error bars. The bar plots can be separated into different colors or facets using metadata labels (for example: plot the average feature values of ‘cohort1’, ‘cohort2’ and ‘cohort3’ in different colors to spot biases).
See also: FeatureDistribution report to plot
the distribution of each feature across examples, rather than only showin the mean value in a bar plot.
Or FeatureComparison report to compare
features across binary metadata labels (e.g., plot the feature value of ‘sick’ repertoires on the x axis,
and ‘healthy’ repertoires on the y axis.).
Example output:
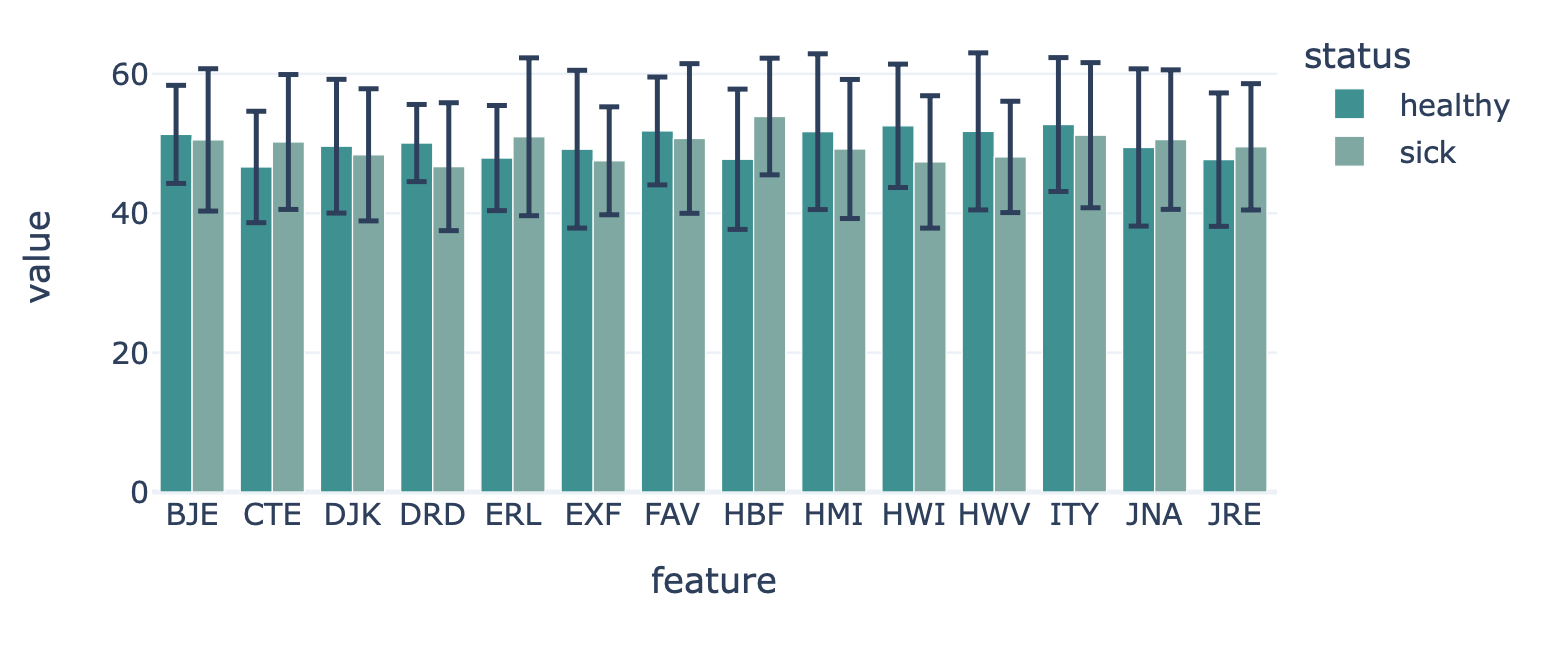
Specification arguments:
color_grouping_label (str): The label that is used to color each bar, at each level of the grouping_label.
row_grouping_label (str): The label that is used to group bars into different row facets.
column_grouping_label (str): The label that is used to group bars into different column facets.
show_error_bar (bool): Whether to show the error bar (standard deviation) for the bars.
x_title (str): x-axis label
y_title (str): y-axis label
plot_top_n (int): plot n of the largest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels. The plot shows averages per label classes.
plot_bottom_n (int): plot n of the smallest features on average separately (useful when there are too many features to plot at the same time). The n features are chosen based on the average feature values across all examples without grouping by labels. The plot shows averages per label classes.
plot_all_features (bool): whether to plot all (might be slow for large number of features)
error_function (str): which error function to use for the error bar. Options are ‘std’ (standard deviation) or ‘sem’ (standard error of the mean). Default: std.
YAML specification:
definitions:
reports:
my_fvb_report:
FeatureValueBarplot: # timepoint, disease_status and age_group are metadata labels
column_grouping_label: timepoint
row_grouping_label: disease_status
color_grouping_label: age_group
plot_all_features: true
plot_top_n: 10
plot_bottom_n: 5
GroundTruthMotifOverlap¶
Creates report displaying overlap between learned motifs and groundtruth motifs implanted in a given sequence dataset. This report must be used in combination with the MotifEncoder.
Specification arguments:
groundtruth_motifs_path (str): Path to a .tsv file containing groundtruth position-specific motifs. The file should specify the motifs as position-specific amino acids, one column representing the positions concatenated with an ‘&’ symbol, the next column specifying the amino acids concatenated with ‘&’ symbol, and the last column specifying the implant rate.
Example:
indices
amino_acids
n_sequences
0
A
4
4&8&9
G&A&C
30
This file shows a motif ‘A’ at position 0 implanted in 4 sequences, and motif G—AC implanted between positions 4 and 9 in 30 sequences
YAML specification:
definitions:
reports:
my_ground_truth_motif_report:
GroundTruthMotifOverlap:
groundtruth_motifs_path: path/to/file.tsv
Matches¶
Reports the number of matches that were found when using one of the following encoders:
MatchedSequences encoder
MatchedReceptors encoder
MatchedRegex encoder
Report results are:
A table containing all matches, where the rows correspond to the Repertoires, and the columns correspond to the objects to match (regular expressions or receptor sequences).
The repertoire sizes (read frequencies and the number of unique sequences per repertoire), for each of the chains. This can be used to calculate the percentage of matched sequences in a repertoire.
When using MatchedSequences encoder or MatchedReceptors encoder, tables describing the chains and receptors (ids, chains, V and J genes and sequences).
When using MatchedReceptors encoder or using MatchedRegex encoder with chain pairs, tables describing the paired matches (where a match was found in both chains) per repertoire.
YAML specification:
definitions:
reports:
my_match_report: Matches
MotifTestSetPerformance¶
This report can be used to show the performance of a learned set motifs using the MotifEncoder
on an independent test set of unseen data.
It is recommended to first run the report MotifGeneralizationAnalysis
in order to calibrate the optimal recall thresholds and plot the performance of motifs on training- and validation sets.
Specification arguments:
test_dataset (dict): parameters for importing a SequenceDataset to use as an independent test set. By default, the import parameters ‘is_repertoire’ and ‘paired’ will be set to False to ensure a SequenceDataset is imported.
YAML specification:
definitions:
reports:
my_motif_report:
MotifTestSetPerformance:
test_dataset:
format: AIRR # choose any valid import format
params:
path: path/to/files/
is_repertoire: False # is_repertoire must be False to import a SequenceDataset
paired: False # paired must be False to import a SequenceDataset
# optional other parameters...
NonMotifSequenceSimilarity¶
Plots the similarity of positions outside the motifs of interest. This report can be used to investigate if the
motifs of interest as determined by the MotifEncoder
have a tendency occur in sequences that are naturally very similar or dissimilar.
For each motif, the subset of sequences containing the motif is selected, and the hamming distances are computed between all sequences in this subset. Finally, a plot is created showing the distribution of hamming distances between the sequences containing the motif. For motifs occurring in sets of very similar sequences, this distribution will lean towards small hamming distances. Likewise, for motifs occurring in a very diverse set of sequences, the distribution will lean towards containing more large hamming distances.
Specification arguments:
motif_color_map (dict): An optional mapping between motif sizes and colors. If no mapping is given, default colors will be chosen.
YAML specification:
definitions:
reports:
my_motif_sim:
NonMotifSimilarity:
motif_color_map:
3: "#66C5CC"
4: "#F6CF71"
5: "#F89C74"
PositionalMotifFrequencies¶
This report must be used in combination with the MotifEncoder.
Plots a stacked bar plot of amino acid occurrence at different indices in any given dataset, along with a plot
investigating motif continuity which displays a bar plot of the gap sizes between the amino acids in the motifs in
the given dataset. Note that a distance of 1 means that the amino acids are continuous (next to each other).
Specification arguments:
motif_color_map (dict): Optional mapping between motif lengths and specific colors to be used. Example:
- motif_color_map:
1: #66C5CC 2: #F6CF71 3: #F89C74
YAML specification:
definitions:
reports:
my_pos_motif_report:
PositionalMotifFrequencies:
motif_color_map:
RelevantSequenceExporter¶
Exports the sequences that are extracted as label-associated when using the SequenceAbundanceEncoder or
CompAIRRSequenceAbundanceEncoder in AIRR-compliant format.
YAML specification:
definitions:
reports:
my_relevant_sequences: RelevantSequenceExporter
ML model reports¶
ML model reports show some type of features or statistics about a single trained ML model.
In the TrainMLModel instruction, ML model reports can be specified inside the ‘selection’ or ‘assessment’ specification under the key ‘reports/models’. Example:
my_instruction:
type: TrainMLModel
selection:
reports:
models:
- my_ml_report
# other parameters...
assessment:
reports:
models:
- my_ml_report
# other parameters...
# other parameters...
BinaryFeaturePrecisionRecall¶
Plots the precision and recall scores for each added feature to the collection of features selected by the BinaryFeatureClassifier.
YAML specification:
definitions:
reports:
my_report: BinaryFeaturePrecisionRecall
Coefficients¶
A report that plots the coefficients for a given ML method in a barplot. Can be used for LogisticRegression, SVM, SVC, and RandomForestClassifier. In the case of RandomForest, the feature importances will be plotted.
When used in TrainMLModel instruction, the report can be specified under ‘models’, both on the selection and assessment levels.
Which coefficients should be plotted (for example: only nonzero, above a certain threshold, …) can be specified. Multiple options can be specified simultaneously. By default the 25 largest coefficients are plotted. The full set of coefficients will also be exported as a csv file.
Example output:
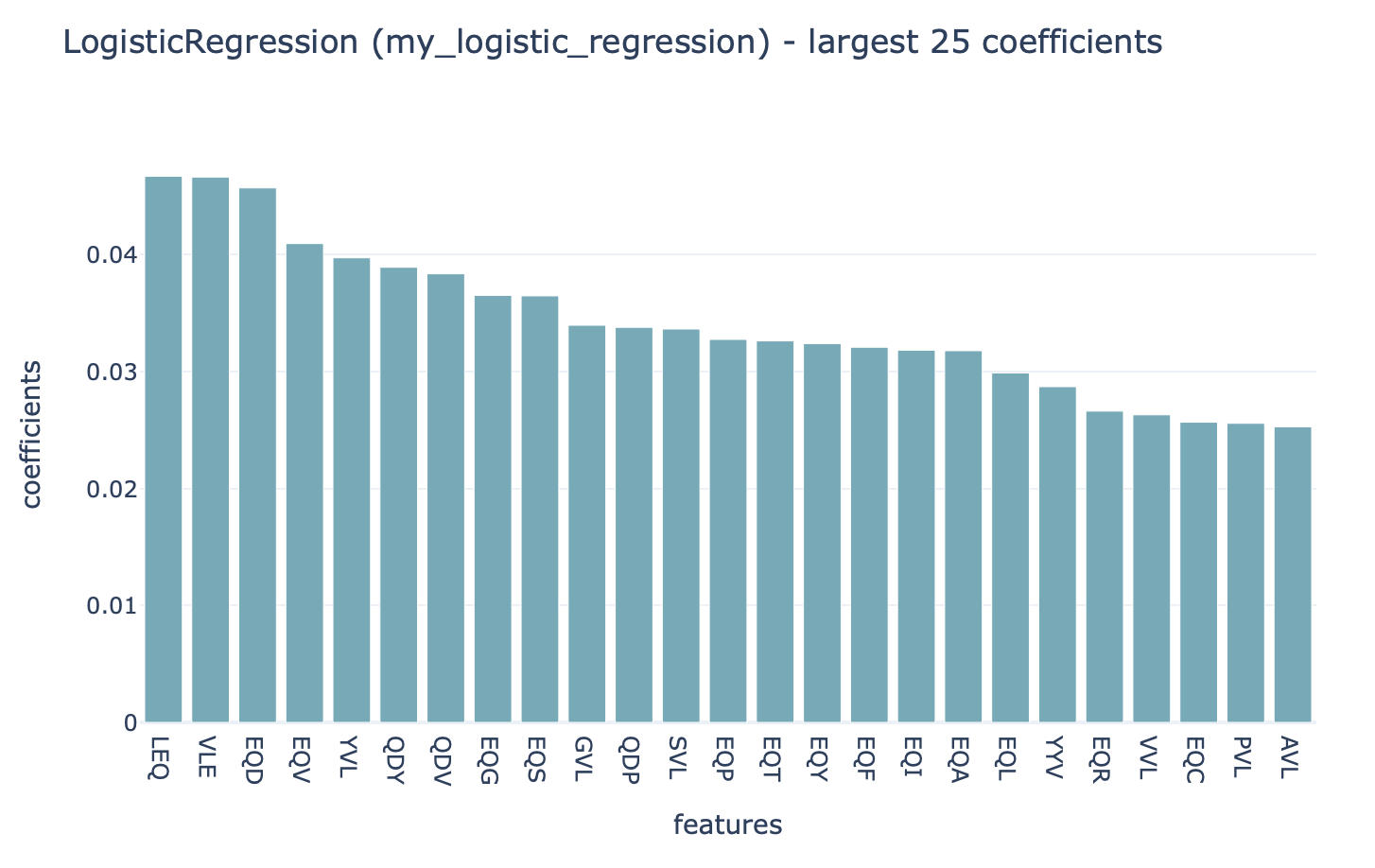
Specification arguments:
coefs_to_plot (list): A list specifying which coefficients should be plotted. Valid values are: ALL, NONZERO, CUTOFF, N_LARGEST.
cutoff (list): If ‘cutoff’ is specified under ‘coefs_to_plot’, the cutoff values can be specified here. The coefficients which have an absolute value equal to or greater than the cutoff will be plotted.
n_largest (list): If ‘n_largest’ is specified under ‘coefs_to_plot’, the values for n can be specified here. These should be integer values. The n largest coefficients are determined based on their absolute values.
YAML specification:
definitions:
reports:
my_coef_report:
Coefficients:
coefs_to_plot:
- all
- nonzero
- cutoff
- n_largest
cutoff:
- 0.1
- 0.01
n_largest:
- 5
- 10
ConfounderAnalysis¶
A report that plots the numbers of false positives and false negatives with respect to each value of the metadata features specified by the user. This allows checking whether a given machine learning model makes more misclassifications for some values of a metadata feature than for the others.
Specification arguments:
metadata_labels (list): A list of the metadata features to use as a basis for the calculations
YAML specification:
definitions:
reports:
my_confounder_report:
ConfounderAnalysis:
metadata_labels:
- age
- sex
ConfusionMatrix¶
A report that plots the confusion matrix for a trained ML method. Supports both binary and multiclass classification.
Specification arguments:
alternative_label (str): optionally, the confusion matrix can be split between different values of an alternative label. This may be useful to compare performance across different data subsets (e.g., batches, sources). If specified, separate confusion matrices will be generated for each value of the alternative label. Default is None.
YAML specification:
definitions:
reports:
my_conf_mat_report: ConfusionMatrix
DeepRCMotifDiscovery¶
This report plots the contributions of (i) input sequences and (ii) kernels to trained DeepRC model with respect to the test dataset. Contributions are computed using integrated gradients (IG). This report produces two figures:
inputs_integrated_gradients: Shows the contributions of the characters within the input sequences (test dataset) that was most important for immune status prediction of the repertoire. IG is only applied to sequences of positive class repertoires.
kernel_integrated_gradients: Shows the 1D CNN kernels with the highest contribution over all positions and amino acids.
For both inputs and kernels: Larger characters in the extracted motifs indicate higher contribution, with blue indicating positive contribution and red indicating negative contribution towards the prediction of the immune status. For kernels only: contributions to positional encoding are indicated by < (beginning of sequence), ∧ (center of sequence), and > (end of sequence).
See DeepRCMotifDiscovery for repertoire classification for a more detailed example.
Reference:
Widrich, M., et al. (2020). Modern Hopfield Networks and Attention for Immune Repertoire Classification. Advances in Neural Information Processing Systems, 33. https://proceedings.neurips.cc//paper/2020/hash/da4902cb0bc38210839714ebdcf0efc3-Abstract.html
Example output:
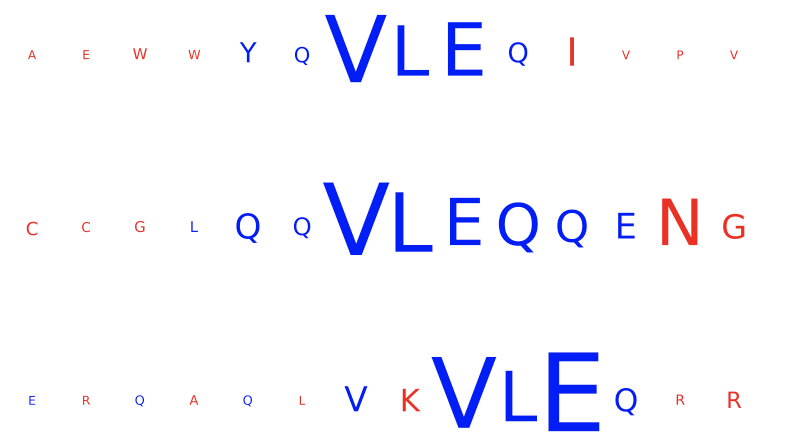

Specification arguments:
n_steps (int): Number of IG steps (more steps -> better path integral -> finer contribution values). 50 is usually good enough.
threshold (float): Only applies to the plotting of kernels. Contributions are normalized to range [0, 1], and only kernels with normalized contributions above threshold are plotted.
YAML specification:
definitions:
reports:
my_deeprc_report:
DeepRCMotifDiscovery:
threshold: 0.5
n_steps: 50
KernelSequenceLogo¶
A report that plots kernels of a CNN model as sequence logos. It works only with trained ReceptorCNN models which has kernels already normalized to represent information gain matrices. Additionally, it also plots the weights in the final fully-connected layer of the network associated with kernel outputs. For more information on how the model works, see ReceptorCNN.
The kernels are visualized using Logomaker. Original publication: Tareen A, Kinney JB. Logomaker: beautiful sequence logos in Python. Bioinformatics. 2020; 36(7):2272-2274. doi:10.1093/bioinformatics/btz921.
YAML specification:
definitions:
reports:
my_kernel_seq_logo: KernelSequenceLogo
MotifSeedRecovery¶
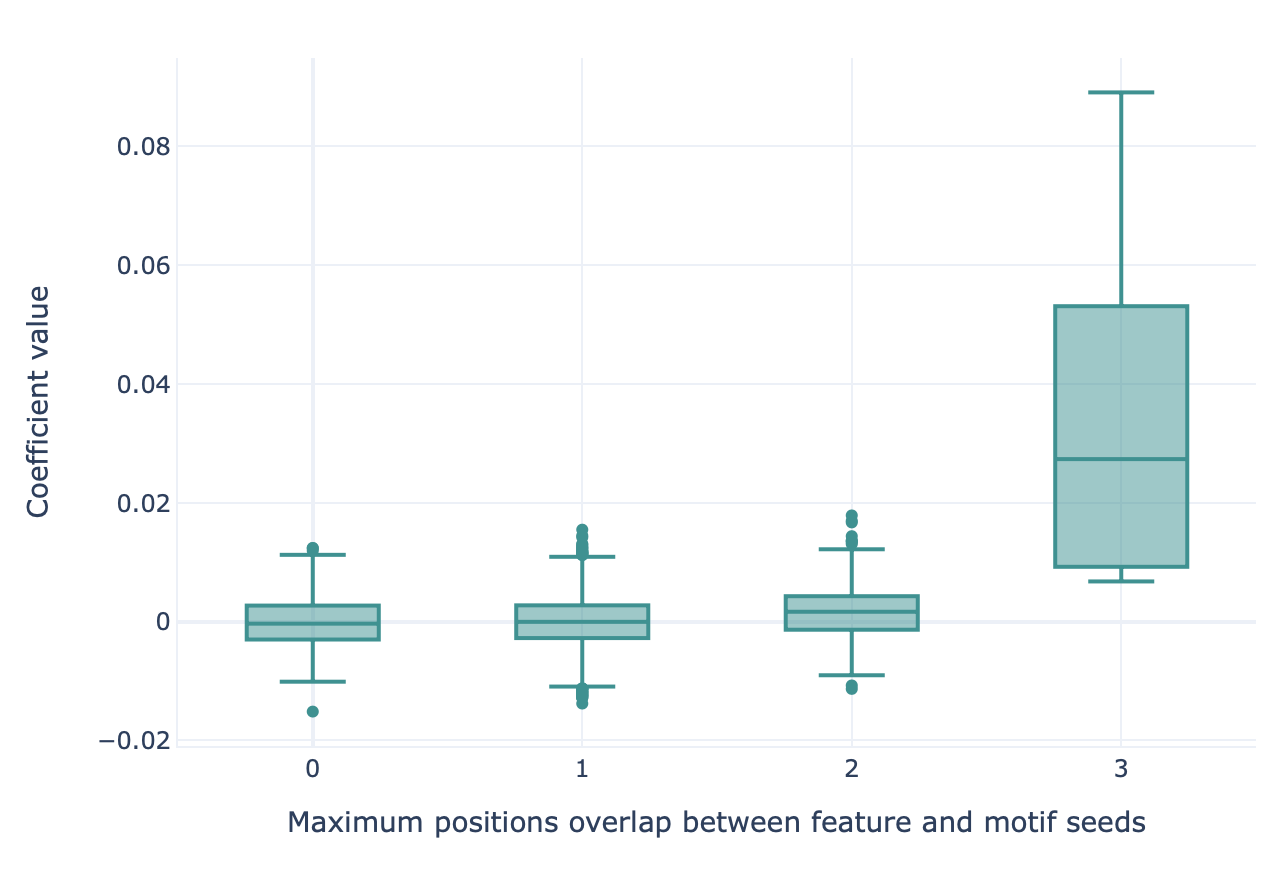
This report can be used to show how well implanted motifs (for example, through the Simulation instruction) can be recovered by various machine learning methods using the k-mer encoding. This report creates a boxplot, where the x axis (box grouping) represents the maximum possible overlap between an implanted motif seed and a kmer feature (measured in number of positions), and the y axis shows the coefficient size of the respective kmer feature. If the machine learning method has learned the implanted motif seeds, the coefficient size is expected to be largest for the kmer features with high overlap to the motif seeds.
Note that to use this report, the following criteria must be met:
KmerFrequencyEncoder must be used.
One of the following classifiers must be used: RandomForestClassifier, LogisticRegression, SVM, SVC
For each label, the implanted motif seeds relevant to that label must be specified
To find the overlap score between kmer features and implanted motif seeds, the two sequences are compared in a sliding window approach, and the maximum overlap is calculated.
Overlap scores between kmer features and implanted motifs are calculated differently based on the Hamming distance that was allowed during implanting.
Without hamming distance:
Seed: AAA -> score = 3
Feature: xAAAx
^^^
Seed: AAA -> score = 0
Feature: xAAxx
With hamming distance:
Seed: AAA -> score = 3
Feature: xAAAx
^^^
Seed: AAA -> score = 2
Feature: xAAxx
^^
Furthermore, gap positions in the motif seed are ignored:
Seed: A/AA -> score = 3
Feature: xAxAAx
^/^^
See Recovering simulated immune signals for more details.
Example output:
Specification arguments:
implanted_motifs_per_label (dict): a nested dictionary that specifies the motif seeds that were implanted in the given dataset. The first level of keys in this dictionary represents the different labels. In the inner dictionary there should be two keys: “seeds” and “hamming_distance”:
seeds: a list of motif seeds. The seeds may contain gaps, specified by a ‘/’ symbol.
hamming_distance: A boolean value that specifies whether hamming distance was allowed when implanting the motif seeds for a given label. Note that this applies to all seeds for this label.
gap_sizes: a list of all the possible gap sizes that were used when implanting a gapped motif seed. When no gapped seeds are used, this value has no effect.
YAML specification:
definitions:
reports:
my_motif_report:
MotifSeedRecovery:
implanted_motifs_per_label:
CD:
seeds:
- AA/A
- AAA
hamming_distance: False
gap_sizes:
- 0
- 1
- 2
T1D:
seeds:
- CC/C
- CCC
hamming_distance: True
gap_sizes:
- 2
ROCCurve¶
A report that plots the ROC curve for a binary classifier.
YAML specification:
definitions:
reports:
my_roc_report: ROCCurve
SequenceAssociationLikelihood¶
Plots the beta distribution used as a prior for class assignment in ProbabilisticBinaryClassifier. The distribution plotted shows the probability that a sequence is associated with a given class for a label.
YAML specification:
definitions:
reports:
my_sequence_assoc_report: SequenceAssociationLikelihood
TCRdistMotifDiscovery¶
The report for discovering motifs in paired immune receptor data of given specificity based on TCRdist3. The receptors are hierarchically clustered based on the tcrdist distance and then motifs are discovered for each cluster. The report outputs logo plots for the motifs along with the raw data used for plotting in csv format.
For the implementation, TCRdist3 library was used (source code available here). More details on the functionality used for this report are available here.
Original publications:
Dash P, Fiore-Gartland AJ, Hertz T, et al. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017; 547(7661):89-93. doi:10.1038/nature22383
Mayer-Blackwell K, Schattgen S, Cohen-Lavi L, et al. TCR meta-clonotypes for biomarker discovery with tcrdist3: quantification of public, HLA-restricted TCR biomarkers of SARS-CoV-2 infection. bioRxiv. Published online December 26, 2020:2020.12.24.424260. doi:10.1101/2020.12.24.424260
Example output:


Specification arguments:
positive_class_name (str): the class value (e.g., epitope) used to select only the receptors that are specific to the given epitope so that only those sequences are used to infer motifs; the reference receptors as required by TCRdist will be the ones from the dataset that have different or no epitope specified in their metadata; if the labels are available only on the epitope level (e.g., label is “AVFDRKSDAK” and classes are True and False), then here it should be specified that only the receptors with value “True” for label “AVFDRKSDAK” should be used; there is no default value for this argument
cores (int): number of processes to use for the computation of the distance and motifs
min_cluster_size (int): the minimum size of the cluster to discover the motifs for
use_reference_sequences (bool): when showing motifs, this parameter defines if reference sequences should be provided as well as a background
YAML specification:
definitions:
reports:
my_tcr_dist_report: # user-defined name
TCRdistMotifDiscovery:
positive_class_name: True # class name, could also be epitope name, depending on how it's defined in the dataset
cores: 4
min_cluster_size: 30
use_reference_sequences: False
TrainingPerformance¶
A report that plots the evaluation metrics for the performance given machine learning model and training dataset.
The available metrics are accuracy, balanced_accuracy, confusion_matrix, f1_micro, f1_macro, f1_weighted, precision,
recall, auc and log_loss (see immuneML.environment.Metric.Metric).
Specification arguments:
metrics (list): A list of metrics used to evaluate training performance. See
immuneML.environment.Metric.Metricfor available options.
YAML specification:
definitions:
reports:
my_performance_report:
TrainingPerformance:
metrics:
- accuracy
- balanced_accuracy
- confusion_matrix
- f1_micro
- f1_macro
- f1_weighted
- precision
- recall
- auc
- log_loss
Clustering method reports¶
Clustering method reports show some features or statistics about the clustering method.
ClusteringVisualization¶
A report that creates low-dimensional visualizations of clustering results using the specified dimensionality reduction method. For each dataset and clustering configuration, it creates a scatter plot where points are colored by their cluster assignments.
Specification arguments:
dim_red_method (dict): specification of which dimensionality reduction to perform; valid options are presented under Dimensionality reduction methods and should be specified with the name of the method and its parameters, see the example below; if not specified, the report will use any dimensionality reduced data present in the dataset’s encoded data; if the dataset does not contain dimensionality reduced data, and the encoded data has more than 2 dimensions, the report will be skipped.
YAML specification:
reports:
my_report_with_pca:
ClusteringVisualization:
dim_red_method:
PCA:
n_components: 2
my_report_with_tsne:
ClusteringVisualization:
dim_red_method:
TSNE:
n_components: 2
init: pca
my_report_existing_dim_red:
ClusteringVisualization:
dim_red_method: null
Dendrogram¶
This report generates a dendrogram visualization from the AgglomerativeClustering method and shows the external labels as annotations.
Specification arguments:
labels (list): List of metadata labels to annotate on the dendrogram.
YAML specification:
reports:
my_dendrogram_report:
Dendrogram:
labels:
- disease_status
- age_group
ExternalLabelClusterSummary¶
This report summarizes the number of examples in a cluster with different values of external labels. For each external label, it creates: 1. A contingency table showing the count of examples for each combination of cluster and label value 2. A heatmap visualization of these counts
It can be used in combination with Clustering instruction.
Specification arguments:
external_labels (list): the list of metadata columns in the dataset that should be compared against cluster assignment
YAML specification:
reports:
my_external_label_cluster_summary:
ExternalLabelClusterSummary:
external_labels: [disease, HLA]
Generative model reports¶
Generative model reports show some type of features or statistics about a generative model.
PWMSummary¶
This report provides the summary of the baseline PWM and shows the following:
probabilities of generated sequences having different lengths
PWMs for each length with positive probability
This report takes no input arguments.
YAML specification:
my_pwm_summary: PWMSummary
VAESummary¶
This report provides the summary of the train SimpleVAE and shows the following:
plots of the latent space after applying a dimensionality reduction method to reduce the data to 2 dimensions, highlighted by V and J gene
plots the histogram for each latent dimension
plots loss per epoch
Specification arguments:
dim_dist_cols (int): how many columns to use to plot the histograms of latent dimensions (either this or dim_dist_rows has to be set, or both)
dim_dist_rows (int): how many rows to use to plot the histogram of latent dimensions (either this or dim_dist_cols has to be set, or both)
dim_red_method (dict): which dimensionality reduction method to use along with its parameters; the method has to return 2 dimensions; see Dimensionality reduction methods for available options; default is PCA with 2 components
YAML specification:
definitions:
reports:
my_vae_summary:
VAESummary:
dim_dist_cols: 4
dim_dist_rows: None
dim_red_method:
PCA:
n_components: 2
Train ML model reports¶
Train ML model reports plot general statistics or export data of multiple models simultaneously when running the TrainMLModel instruction.
In the TrainMLModel instruction, train ML model reports can be specified under ‘reports’. Example:
my_instruction:
type: TrainMLModel
reports:
- my_train_ml_model_report
# other parameters...
CVFeaturePerformance¶
This report plots the average training vs test performance w.r.t. given encoding parameter which is explicitly set in the feature attribute. It can be used only in combination with TrainMLModel instruction and can be only specified under ‘reports’
Specification arguments:
feature: name of the encoder parameter w.r.t. which the performance across training and test will be shown. Possible values depend on the encoder on which it is used.
is_feature_axis_categorical (bool): if the x-axis of the plot where features are shown should be categorical; alternatively it is automatically determined based on the feature values
YAML specification:
definitions:
reports:
report1:
CVFeaturePerformance:
feature: p_value_threshold # parameter value of SequenceAbundance encoder
is_feature_axis_categorical: True # show x-axis as categorical
DiseaseAssociatedSequenceCVOverlap¶
DiseaseAssociatedSequenceCVOverlap report makes one heatmap per label showing the overlap of disease-associated sequences (or k-mers)
produced by the SequenceAbundanceEncoder,
CompAIRRSequenceAbundanceEncoder or
KmerAbundanceEncoder
between folds of cross-validation (either inner or outer loop of the nested CV). The overlap is computed by the following equation:
For details, see Greiff V, Menzel U, Miho E, et al. Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Reports. 2017;19(7):1467-1478. doi:10.1016/j.celrep.2017.04.054.
Specification arguments:
compare_in_selection (bool): whether to compute the overlap over the inner loop of the nested CV - the sequence overlap is shown across CV folds for the model chosen as optimal within that selection
compare_in_assessment (bool): whether to compute the overlap over the optimal models in the outer loop of the nested CV
YAML specification:
definitions:
reports:
my_overlap_report:
DiseaseAssociatedSequenceCVOverlap:
compare_in_selection: false
compare_in_assessment: true
MLSettingsPerformance¶
Report for TrainMLModel instruction: plots the performance for each of the setting combinations as defined under ‘settings’ in the assessment (outer validation) loop.
The performances are grouped by label (horizontal panels) encoding (vertical panels) and ML method (bar color). When multiple data splits are used, the average performance over the data splits is shown with an error bar representing the standard deviation.
This report can be used only with TrainMLModel instruction under ‘reports’.
Specification arguments:
single_axis_labels (bool): whether to use single axis labels. Note that using single axis labels makes the figure unsuited for rescaling, as the label position is given in a fixed distance from the axis. By default, single_axis_labels is False, resulting in standard plotly axis labels.
x_label_position (float): if single_axis_labels is True, this should be an integer specifying the x axis label position relative to the x axis. The default value for label_position is -0.1.
y_label_position (float): same as x_label_position, but for the y-axis.
YAML specification:
definitions:
reports:
my_hp_report: MLSettingsPerformance
PerformancePerLabel¶
Report that shows the performance of the model where the examples are grouped by alternative_label. It can be used to investigate if the model is learning the alternative_label instead of label of interest for classification.
Specification arguments:
alternative_label (str): The name of the alternative_label column in the dataset.
metric (str): The metric to use for the report. Default is balanced_accuracy.
compute_for_selection (bool): If True, the report will be computed for the selection. Default is True.
compute_for_assessment (bool): If True, the report will be computed for the assessment. Default is True.
plot_on_test (bool): If True, the report will be plotted on the test data. Default is True.
plot_on_train (bool): If True, the report will be plotted on the training data. Default is False.
YAML specification:
reports:
my_report:
PerformancePerLabel:
alternative_label: batch
metric: balanced_accuracy
PrecisionRecallCurveSummary¶
This report plots Precision-Recall curves for all trained ML settings ([preprocessing], encoding, ML model) in the outer loop of cross-validation in the TrainMLModel instruction. It also reports the average precision (AP) for each setting.
If there are multiple splits in the outer loop, this report will make one
plot per split. This report is defined only for binary classification.
YAML specification:
definitions:
reports:
my_pr_summary_report: PrecisionRecallCurveSummary
ROCCurveSummary¶
This report plots ROC curves for all trained ML settings ([preprocessing], encoding, ML model) in the outer loop of cross-validation in the TrainMLModel instruction. If there are multiple splits in the outer loop, this report will make one plot per split. This report is defined only for binary classification. If there are multiple labels defined in the instruction, each label has to have two classes to be included in this report.
YAML specification:
definitions:
reports:
my_roc_summary_report: ROCCurveSummary
ReferenceSequenceOverlap¶
The ReferenceSequenceOverlap report compares a list of disease-associated sequences (or k-mers) produced by the
SequenceAbundanceEncoder,
CompAIRRSequenceAbundanceEncoder or
KmerAbundanceEncoder to
a list of reference sequences. It outputs a Venn diagram and a list of sequences found both in the encoder and reference list.
The report compares the sequences by their sequence content and the additional comparison_attributes (such as V or J gene), as specified by the user.
Specification arguments:
reference_path (str): path to the reference file in csv format which contains one entry per row and has columns that correspond to the attributes listed under comparison_attributes argument
comparison_attributes (list): list of attributes to use for comparison; all of them have to be present in the reference file where they should be the names of the columns
label (str): name of the label for which the reference sequences/k-mers should be compared to the model; if none, it takes the one label from the instruction; if it is none and multiple labels were specified for the instruction, the report will not be generated
YAML specification:
definitions:
reports:
my_reference_overlap_report:
ReferenceSequenceOverlap:
reference_path: reference_sequences.csv # example usage with SequenceAbundanceEncoder or CompAIRRSequenceAbundanceEncoder
comparison_attributes:
- sequence_aa
- v_call
- j_call
my_reference_overlap_report_with_kmers:
ReferenceSequenceOverlap:
reference_path: reference_kmers.csv # example usage with KmerAbundanceEncoder
comparison_attributes:
- k-mer
Clustering Instruction Reports¶
ExternalLabelMetricHeatmap¶
This report creates heatmaps comparing clustering methods against external labels for each metric. For each external label and metric combination, it creates:
A table showing the mean and standard deviation of metric values across splits for each combination of clustering method and external label
A heatmap visualization where the color represents the mean value and the text shows mean±std
The external labels and metrics are automatically determined from the clustering instruction specification.
YAML specification:
reports:
my_external_label_metric_heatmap: ExternalLabelMetricHeatmap
Multi dataset reports¶
Multi dataset reports are special reports that can be specified when running immuneML with the MultiDatasetBenchmarkTool.
See Manuscript use case 1: Robustness assessment for an example.
When running the MultiDatasetBenchmarkTool, multi dataset reports can be specified under ‘benchmark_reports’.
Example:
my_instruction:
type: TrainMLModel
benchmark_reports:
- my_benchmark_report
# other parameters...
DiseaseAssociatedSequenceOverlap¶
DiseaseAssociatedSequenceOverlap report makes a heatmap showing the overlap of disease-associated sequences (or k-mers)
produced by the SequenceAbundanceEncoder,
CompAIRRSequenceAbundanceEncoder or
KmerAbundanceEncoder
between multiple datasets of different sizes (different number of repertoires per dataset).
This plot can be used only with MultiDatasetBenchmarkTool.
The overlap is computed by the following equation:
For details, see: Greiff V, Menzel U, Miho E, et al. Systems Analysis Reveals High Genetic and Antigen-Driven Predetermination of Antibody Repertoires throughout B Cell Development. Cell Reports. 2017;19(7):1467-1478. doi:10.1016/j.celrep.2017.04.054.
YAML specification:
definitions:
reports:
my_overlap_report: DiseaseAssociatedSequenceOverlap # report has no parameters
PerformanceOverview¶
PerformanceOverview report creates an ROC plot and precision-recall plot for optimal trained models on multiple datasets. The labels on the plots are the names of the datasets, so it might be good to have user-friendly names when defining datasets that are still a combination of letters, numbers and the underscore sign.
This report can be used only with MultiDatasetBenchmarkTool as it will plot ROC and PR curve for trained models across datasets. Also, it requires the task to be immune repertoire classification and cannot be used for receptor or sequence classification. Furthermore, it uses predictions on the test dataset to assess the performance and plot the curves. If the parameter refit_optimal_model is set to True, all data will be used to fit the optimal model, so there will not be a test dataset which can be used to assess performance and the report will not be generated.
If datasets have the same number of examples, the baseline PR curve will be plotted as described in this publication: Saito T, Rehmsmeier M. The Precision-Recall Plot Is More Informative than the ROC Plot When Evaluating Binary Classifiers on Imbalanced Datasets. PLOS ONE. 2015;10(3):e0118432. doi:10.1371/journal.pone.0118432
If the datasets have different number of examples, the baseline PR curve will not be plotted.
YAML specification:
definitions:
reports:
my_performance_report: PerformanceOverview